
碎片印迹聚合物的制备及其对薯蓣皂苷的分子识别
2023-03-04 01:23肖真仪杨城城刘美君周小辉彭雨婷简宇轩
湖南师范大学自然科学学报 2023年6期
肖真仪,邓 文,杨城城,刘美君,周小辉,彭雨婷,简宇轩,李 辉
(吉首大学化学化工学院,中国 吉首 416000)
由于环境及生物样品的复杂性,常规分离方法难以高效完成目标药物及生物质的分离检测,迫切需要高选择性的分离方法[1-3]。分子印迹技术在分析领域无疑极具吸引力,因为这种技术创造了选择性结合位点,将目标分子的特征通过印迹而保留[4-6]。但该项技术仍有一些局限性,如高毒物质不能用作模板制备印迹材料[7,8],模板泄露则导致无法正确定量测定及模板净化[9,10]。碎片印迹技术不是采用目标分子作模板,而是使用目标化合物的部分结构或片段作为模板制备印迹聚合物,这样就可消除分子识别中的模板残留[11-14]。
薯蓣皂苷(Dioscin)是一种具有广阔生物活性的天然产物[15],常存在于一些药用植物的根茎中,具有抗骨质疏松、抗癌、抗炎、抗神经变性、抗感染及抗衰老[16-19]等药理活性,对心血管疾病、过敏性疾病、2型糖尿病、神经退行性疾病、皮肤老化及更年期症状有较好的预防和治疗效果[20-22]。从植物中提取并分离薯蓣皂苷是获取该化合物的主要来源,但薯蓣皂苷水溶性差[23]、生物利用度低[24,25],大大限制了薯蓣皂苷的提取和应用。基于分子印迹材料的高选择吸附性能,将这种具有特异识别能力的印迹聚合物作为固相萃取吸附剂以实现对薯蓣皂苷的分离和富集是一种低成本、高效率且操作简单的分离分析技术。本工作以葡萄糖及齐墩果酸为模板碎片,采用沉淀法制备印迹聚合物,探讨这种碎片印迹聚合物对目标化合物薯蓣皂苷的吸附及提取分离效能。
1 实验部分
1.1 试剂及材料
薯蓣皂苷(99%,质量分数,下同)、4-乙烯基吡啶(98%)、N-异丙基丙烯酰胺(98%)及交联剂乙二醇二甲基丙烯酸酯(98%)均购自河南省万家标准物质研发中心有限公司;偶氮二异丁腈(98%)购自美国阿拉丁工业公司;色谱纯的正硅酸乙酯、乙腈及甲醇购自成都金山化学试剂有限公司;分析纯的乙酸、乙醇及甲苯购自天津市永大化学试剂有限公司。葡萄糖(98%)和齐墩果酸(97%)购自中国医药生物制品检定所。
薯蓣皂苷样品溶液:称取20 g干燥的盾叶薯蓣茎[26],粉碎,浸入200 mL含30%盐酸的乙醇-水溶液(体积比7∶3)中,超声处理2 h,过滤,滤渣再用同样的溶剂重复提取2次,合并提取液。减压蒸馏除去大部分溶剂后,用少量乙酸-乙腈混合溶液(体积比2∶8)溶解,过滤除去固体物,所得溶液即为薯蓣皂苷样品溶液,备用。
1.2 设备及仪器
S-3400N扫描电镜(日本);WGH-30A 傅里叶红外光谱仪(中国天津仪器有限公司);LC-40D 高效液相色谱仪和UV-2550 紫外可见光谱仪(日本岛津公司);TD-3500 X射线衍射仪(上海第二光学仪器厂);高速台式离心机TGL-16(金坛大地仪器厂);电子天平FA2104N(上海菁海仪器公司);真空干燥箱DZ-1AII(天津泰斯特仪器公司);超声波清洗器KQ250E(昆山市超声仪器厂);恒温水浴锅GSY-Ⅱ(北京市永光明医疗仪器厂)。
1.3 薯蓣皂苷印迹聚合物的制备
将一定量的模板分子加入到10 mL甲苯-甲醇(体积比为4∶1)混合溶剂中,超声处理5 min,加入0.3 mmol 4-乙烯基吡啶、0.3 mmol N-异丙基丙烯酰胺、10.0 mmol乙二醇二甲基丙烯酸酯及20.0 mg偶氮二异丁腈。再用超声波处理5 min,通入氮气处理15 min,密封,反应体系置于333 K油浴锅中反应24 h后,将混合物加入到50.0 mL 20%乙酸-甲醇混合溶液中,静置4 h后,除去上层清液,再用同样体积的乙酸-甲醇混合溶液重复浸泡2次,过滤,固体置于真空干燥箱中干燥12 h,即得薯蓣皂苷印迹聚合物。制备非印迹聚合物(NIP)时,除不加模板分子外,其他步骤与印迹聚合物的制备步骤相同。各印迹聚合物模板的类型及用量参见表1。当用葡萄糖或齐墩果酸为模板时,分子印迹聚合物的命名为GL-MIP或OL-MIP。
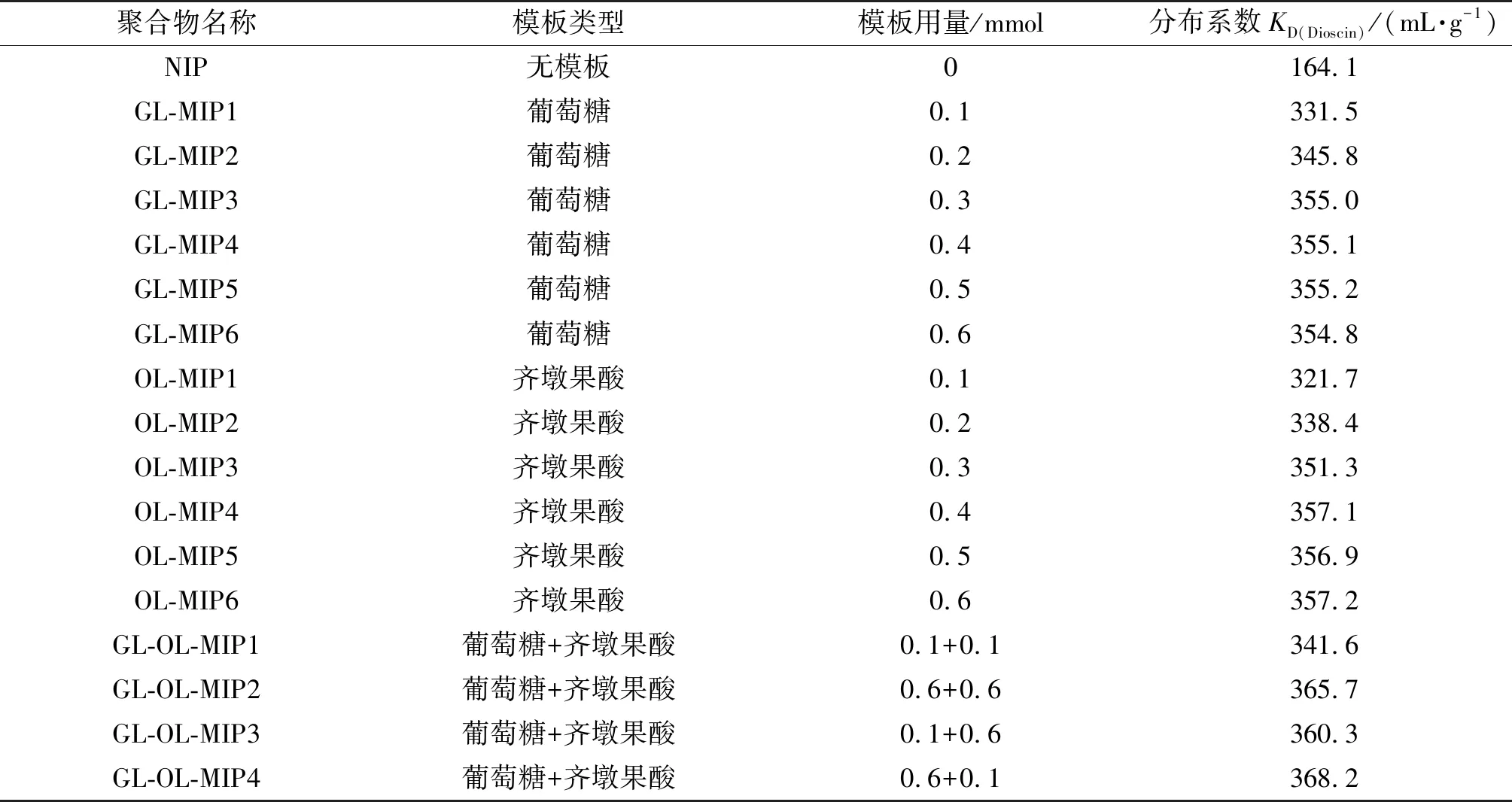
表1 碎片印迹聚合物的制备a
1.4 红外光谱分析
样品置于373 K真空干燥箱中干燥4 h,然后测试分子印迹聚合物和非印迹聚合物的红外光谱图。
1.5 电镜分析
测试分子印迹聚合物和非印迹聚合物的电镜图,观察颗粒大小和表面形貌。
1.6 吸附测试
1.6.1 吸附动力学 在20 mL 0.10 g·L-1薯蓣皂苷的乙腈溶液中,加入20 mg分子印迹聚合物,吸附开始后,每间隔0.5 h后,取少量上清液,用HPLC测定上清液中薯蓣皂苷的质量浓度,按方程(1)计算不同时间下分子印迹聚合物的吸附量Qt(mg·g-1)。平行测定3次,取其平均值。
(1)
式中,c0和ct分别为薯蓣皂苷的初始质量浓度和吸附t时间后的质量浓度,g·L-1;V为溶液的体积,mL;m为吸附剂的质量,g。
1.6.2 吸附等温线 在5.0 mL浓度不同的薯蓣皂苷-乙腈溶液中,分别加入5 mg分子印迹聚合物,吸附4 h后,用HPLC法测定各溶液中薯蓣皂苷浓度。按方程(2)—(4)计算(Qe,mg/g)、分布系数KD(mL·g-1)及选择因子α。
(2)
(3)
(4)
其中c0和ce分别为薯蓣皂苷的初始浓度和平衡浓度,g·L-1;V为溶剂体积,mL;m为吸附剂质量, g。KD(template)和KD(analogue)分别为模板及类似物的分布系数。
1.7 分子印迹固相萃取
1.7.1 萃取柱的制备 取一根底端装有筛板的萃取用空柱管,用甲醇和去离子水洗净,晾干,称取1.5 g薯蓣皂苷印迹聚合物,均匀装填到柱中,在装填过程中边装填边轻轻敲打柱管,使聚合物填充均匀、紧实。然后,依次用10 mL甲醇及10 mL乙腈溶液流过固相萃取柱,去除聚合物中杂质,使萃取柱活化,备用。
1.7.2 上样和洗脱 取3.0 mL样品溶液上样,下端用真空抽吸,收集流出液。上样后依次用2×1 mL 乙腈、2×1 mL 乙酸乙酯、5×1 mL H2O-C2H5OH(体积比1∶1)混合液、5×1 mL H2O-CH3OH(体积比1∶1)混合液、5×1 mL甲醇、3×1 mL甲醇-乙酸(体积比8∶2)进行洗涤和洗脱,分别收集各淋洗与洗脱步骤的流出液,进行HPLC分析。
1.8 高效液相色谱分析
高效液相色谱分析在C18柱上进行,以乙腈-水(体积比9∶1)混合溶剂为流动相,流速为1.0 mL·min-1,检测波长203 nm,进样体积为10 μL。采用标准曲线法进行定性和定量。薯蓣皂苷标准曲线方程为:
Y=9 332.3c-12.24,R2=0.999 0。
式中:Y为峰面积;c为底物浓度,g ·L-1;R2为相关系数。
2 结果与讨论
2.1 分子印迹聚合物的制备和表征
根据薯蓣皂苷的分子结构(如图1所示),葡萄糖及齐墩果酸为目标分子中的部分结构或具有部分相似的结构,以这两种碎片分子作为模板,采用沉淀聚合法制备了分子印迹聚合物。4-乙烯基吡啶和N-异丙基丙烯酰胺为混合功能单体,二者均可与模板分子中的羟基形成氢键复合物。为了考察碎片印迹聚合物对目标化合物薯蓣皂苷的分子识别能力,在制备时,其他单体及用量与制备条件均固定不变,仅改变模板的类型及用量。

图1 几种化合物的分子结构
分别以葡萄糖或齐墩果酸为模板,考察不同模板用量下分子印迹聚合物的分布系数KD (Dioscin)。为便于比较,将非印迹聚合物的分布系数也列入其中。从表1中的结果可以看出,无论是以葡萄糖还是齐墩果酸为模板,也不管模板用量的多少,制备的分子印迹聚合物的分布系数明显高于非印迹聚合物。这表明以葡萄糖或齐墩果酸为模板时,在印迹聚合物基体中形成了可对薯蓣皂苷具备选择作用的识别位点。在制备过程中,当模板分子葡萄糖或齐墩果酸的用量增加时,分子印迹材料的分布系数增加,但当葡萄糖用量超过0.3 mmol,齐墩果酸用量超过0.4 mmol时,分子印迹聚合物的分布系数变化不大。显然,当模板用量过低时,在聚合物基体中形成的印迹位点过少,不利于印迹聚合物的分子吸附。但印迹聚合物的分布系数也并非完全与模板用量成正比,这可能是由于当模板浓度较高时,易形成模板分子低聚物,从而改变印迹空穴的大小和形状,引起印迹空穴-薯蓣皂苷分子间的适应性降低所致。由于葡萄糖分子及齐墩果酸分子均含有与目标化合物薯蓣皂苷分子相似的分子碎片,于是,使用二者的混合物为模板制备印迹材料。采用因子设计,考察在不同葡萄糖用量及齐墩果酸用量间的交互作用下,所得分子印迹材料的分布系数。结果显示,在齐墩果酸用量固定时,印迹材料的分布系数与葡萄糖用量呈现了一定的正相关性。当二者用量均为0.6 mmol时,所得印迹材料的分布系数并非最高,而当葡萄糖和齐墩果酸的用量分别为0.6和0.1 mmol时,分子印迹聚合物(GL-OL-MIP4)的分布系数最大,其值为368.2 mL·g-1。采用混合碎片作为模板,由于此方式增加了识别位点类型,有助于提高印迹材料的吸附性能。但值得注意的是,这种交互作用也显示了两种碎片对印迹材料的分子识别具有不同程度的影响。

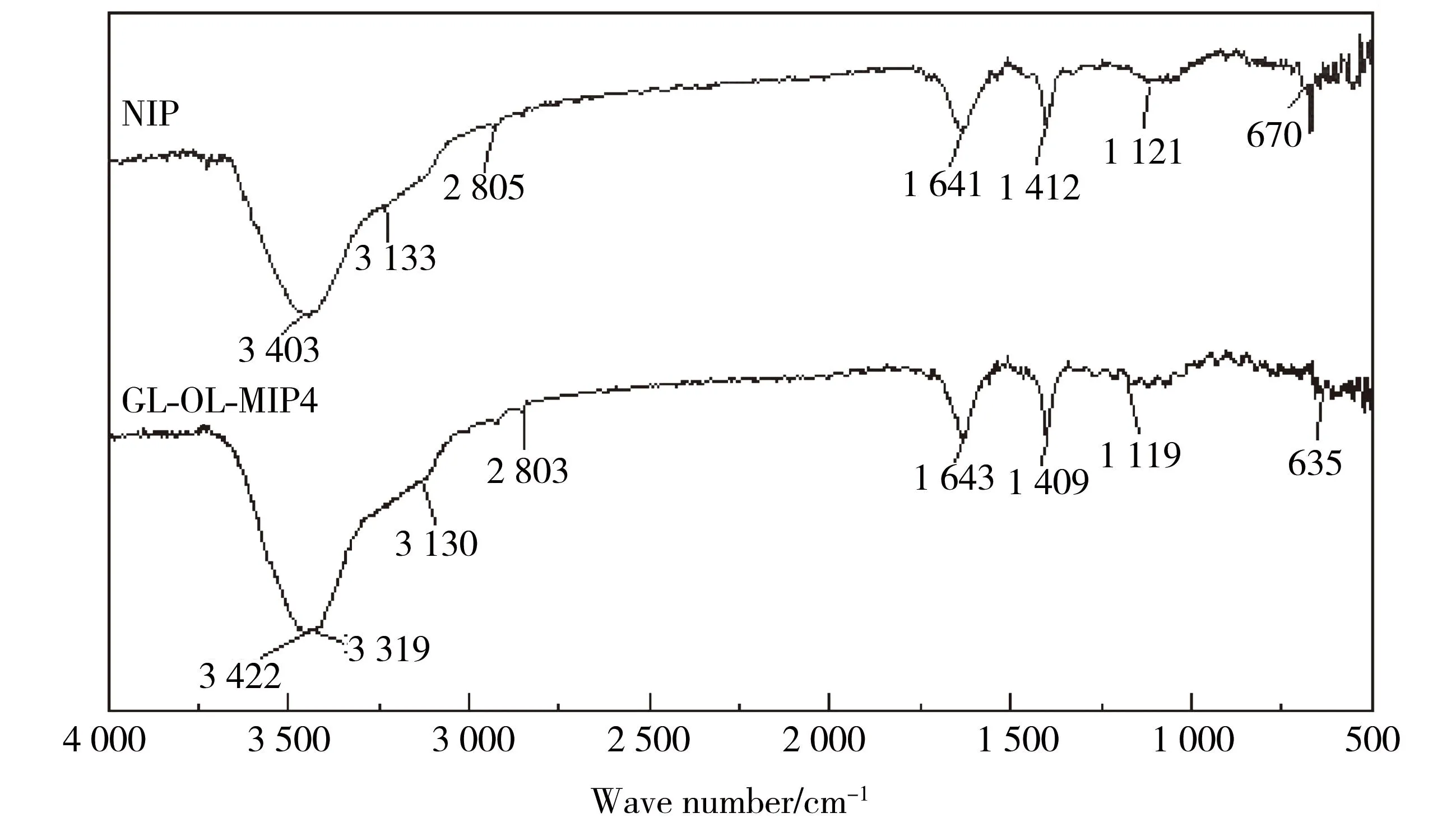
图2 分子印迹聚合物(GL-OL-MIP4)及非印迹聚合物(NIP)的红外光谱

图3 分子印迹聚合物(GL-OL-MIP4)及非印迹聚合物(NIP)的扫描电镜图
2.2 分子印迹聚合物的吸附性能
2.2.1 等温吸附 设定温度为30 ℃,测试分子印迹聚合物(GL-OL-MIP4)对目标化合物薯蓣皂苷的等温吸附能力。如图4所示,碎片印迹聚合物对薯蓣皂苷具有较强的吸附能力,其饱和吸附量约为34.67 mg·g-1,远高于非印迹材料NIP(其饱和吸附量约为14.59 mg·g-1)。当底物浓度相同时,印迹材料的吸附能力均强于非印迹聚合物,这可能源于碎片分子的印迹孔穴对目标化合物有分子识别作用。这也显示了目标分子碎片在分子印迹识别应用中的可能性。

图4 分子印迹聚合物(GL-OL-MIP4)的吸附等温线
为了探讨碎片印迹材料中结合位点特征,对所得碎片印迹聚合物的吸附等温值进行Scatchard分析,以研究其结合位点分布。采用方程(5)对等温吸附数据进行线性拟合,得到如图5所示的直线图,其中分子印迹聚合物(GL-OL-MIP4)的Scatchard分析线图表明该印迹基体中主要存在两类吸附位点,根据直线的斜率和截距可以计算出最大表观位点数目(Qmax)及平衡离解常数(Kd)。而非印迹聚合物基体中主要存在一类吸附位点。表2给出了印迹及非印迹材料基体中各类吸附位点的Qmax和Kd。对于GL-OL-MIP4印迹聚合物基体中的高亲和位点,Qmax和Kd分别为1.762×10-4mol·g-1和1.970×10-4mol·L-1,而低亲和位点,其值分别为9.441×10-5mol·g-1和3.782×10-5mol·L-1;对于NIP,Qmax和Kd分别为5.501×10-5mol·g-1和1.763×10-4mol·L-1。

表2 吸附等温值的Scatchard分析
(5)
2.2.2 吸附选择性 考察印迹材料对模板薯蓣皂苷及相关化合物薯蓣皂苷元、重楼皂苷及重楼皂苷元(分子结构示于图1)的静态吸附,测试碎片印迹聚合物GL-OL-MIP4的吸附选择性,得到表3。薯蓣皂苷提取物中常含有这些化合物,测定印迹聚合物对目标的选择性可为提取分离目标化合物提供基础。从表中结果可以看出,印迹材料对目标化合物薯蓣皂苷具有一定的选择性,最高选择因子为1.356。选择性不够高的原因在于这些相关化合物的分子结构中都含有与碎片模板的类似结构。但印迹材料仍对目标化合物具有较高的吸附选择性。
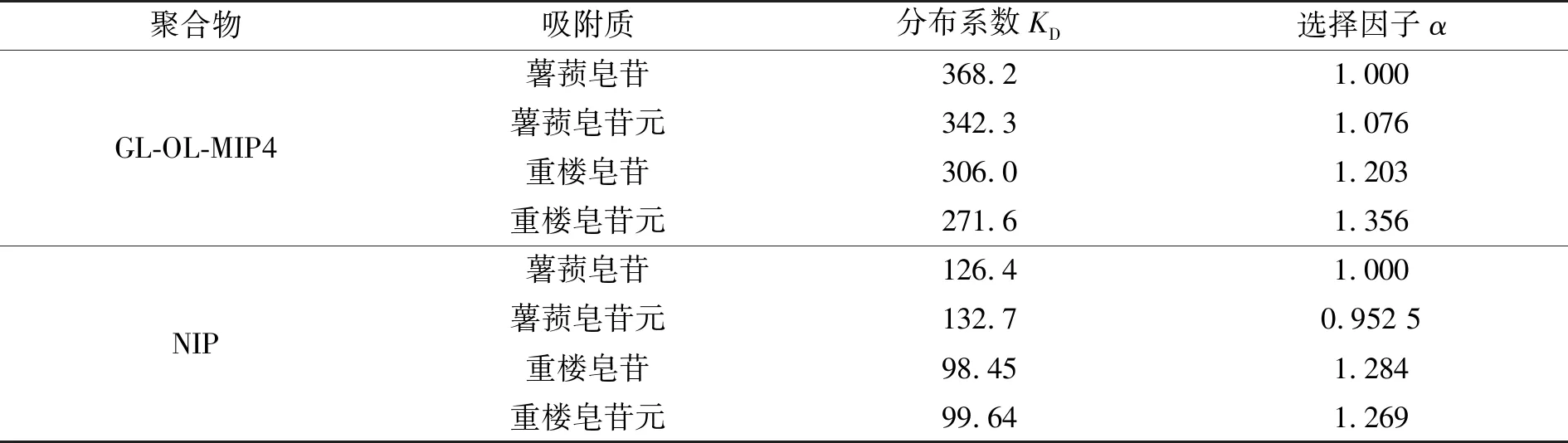
表3 分子印迹聚合物(GL-OL-MIP4)对目标化合物薯蓣皂苷的选择性
2.3 分子印迹固相萃取
2.3.1 模拟样品溶液的固相萃取 以分子印迹聚合物GL-OL-MIP4为固相萃取吸附剂,考察其对含0.1 g·L-1的薯蓣皂苷、薯蓣皂苷元、重楼皂苷及重楼皂苷元模拟混合溶液中目标化合物薯蓣皂苷的提取分离效能。取一定体积(3.0 mL)的模拟样品溶液上样,收集流出液,测定流出液中薯蓣皂苷浓度,计算装载量。依次用2×1 mL 乙腈、2×1 mL 乙酸乙酯、5×1 mL H2O-C2H5OH(体积比1∶1,下同)混合液、5×1 mL H2O-CH3OH(1∶1)混合液、5×1 mL甲醇、3×1 mL甲醇-乙酸(8∶2)进行洗涤和洗脱,提取过程中,分别收集各淋洗与洗脱步骤的流出液,而后进行HPLC分析,计算各步骤中薯蓣皂苷回收率。结果如表4所示。可以发现,当使用分子印迹聚合物萃取时,用2×1 mL 乙腈和2×1 mL乙酸乙酯进行洗涤,主要洗涤杂质化合物,仅有少量的目标化合物脱出。而用5×1 mL H2O-C2H5OH(1∶1)混合液和5×1 mL H2O-CH3OH(1∶1)混合液进行洗脱时,可以回收大部分目标化合物(薯蓣皂苷回收率达86.01%),薯蓣皂苷得到了有效富集和分离。而非印迹聚合物富集和分离效能较低。另外,分子印迹固相萃取的总回收率达99.66%,显示了较好应用效能。
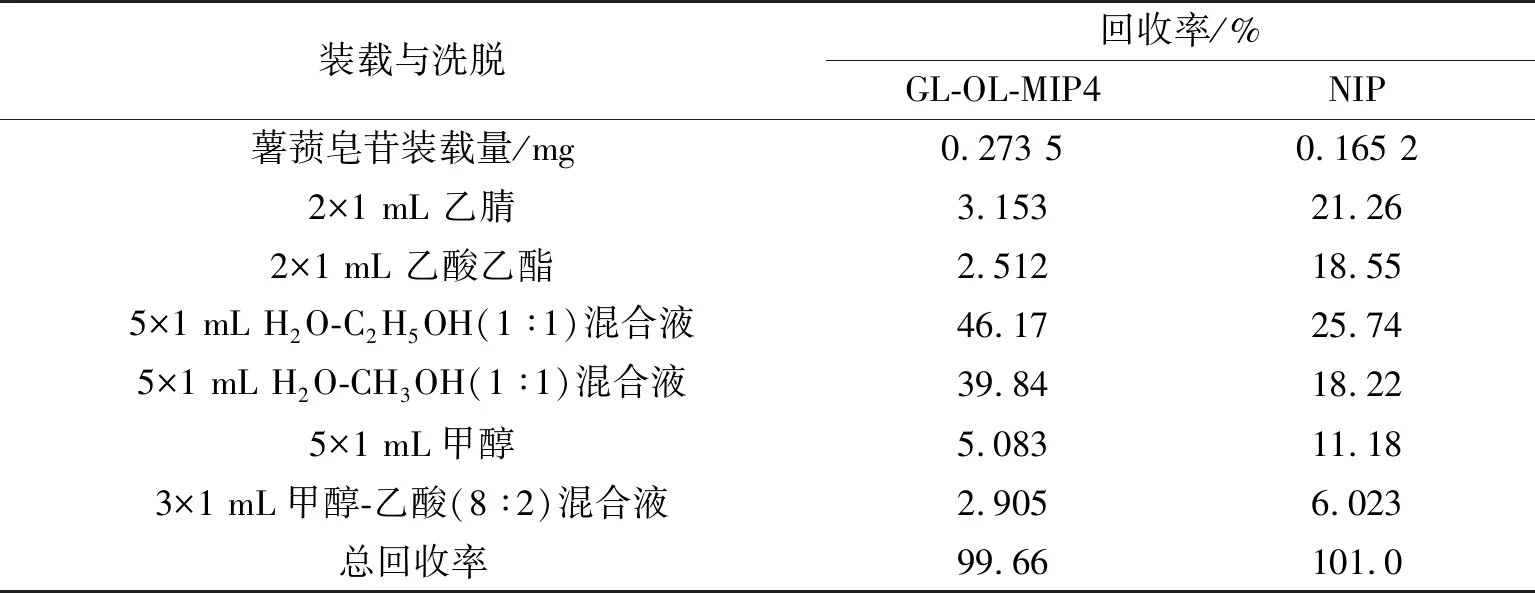
表4 分子印迹聚合物固相萃取
2.3.2 实际样品的固相萃取 取5.0 mL的样品溶液上样,收集流出液,静置3 min后,按照模拟样品溶液的萃取方式,依次用2×1 mL 乙腈、2×1 mL 乙酸乙酯、5×1 mL H2O-C2H5OH(1∶1)混合液、5×1 mL H2O-CH3OH(1∶1)混合液、5×1 mL甲醇、3×1 mL甲醇-乙酸(8∶2)进行洗涤和洗脱,提取过程中,分别收集用5×1 mL H2O-C2H5OH(1∶1)和5×1 mL H2O-CH3OH(1∶1)混合液洗脱时的流出液,合并流出液,减压蒸馏除去大部分溶剂后,再用少量乙醇溶解,HPLC法测定产品中薯蓣皂苷含量。图6给出了样品溶液及分子印迹固相萃取提取液的高效液相色谱图。保留时间为17.85 min的色谱峰为薯蓣皂苷。可以发现,对于成分较为复杂的样品溶液(显示多色谱峰,见图6a)经分子印迹固相萃取和分离后,产品中杂质峰大幅减少(图6b)。粗提物中的薯蓣皂苷得到了较好的分离和纯化,显示了分子印迹聚合物提取样品溶液中的薯蓣皂苷时,具有较好的提取分离应用效能。

图6 盾叶薯蓣粗提液及分子印迹固相萃取产物的HPLC图
2.4 重现性测试
为了测定分子印迹聚合物的重复使用性能。将1.0 g分子印迹聚合物加入到5.0 mL质量浓度为0.6 g·L-1的薯蓣皂素标准溶液中,吸附4 h后,过滤,计算吸附量。固体用甲醇-乙酸(9∶1)混合溶液洗净吸附在聚合物上的目标化合物,过滤后将固体重新加入到5 mL等浓度的模板溶液中,吸附、过滤,测定吸附量。重复操作10次。每次使用后,印迹聚合物的吸附量标示于图7中。结果发现吸附量改变不大,可以重复使用。

图7 分子印迹聚合物使用重现性
3 结论
以葡萄糖及齐墩果酸为模板碎片,4-乙烯基吡啶-N-异丙基丙烯酰胺为共同功能单体,制备了一种可用于选择提取薯蓣皂苷的碎片印迹微球。当两种模板碎片(葡萄糖和齐墩果酸)用量分别为0.6和0.1 mmol时,所得印迹聚合物GL-OL-MIP4的分布系数最大(其值为368. 2 mL·g-1)。该印迹微球平均粒径约11.3 μm,对薯蓣皂苷具有较强的吸附能力,饱和吸附量约34.67 mg·g-1。Scatchard分析表明这种碎片印迹材料(GL-OL-MIP4)主要有两类亲和位点,其中,高亲和位点的表观键合位点数目Qmax和吸附离解常数Kd分别为1.762×10-4mol·g-1和1.970×10-4mol·L-1;对于低亲和位点,其值分别为9.441×10-5mol·g-1和3.782×10-5mol·L-1。印迹材料对目标化合物薯蓣皂苷具有一定的选择性,选择因子为1.356。当使用该印迹聚合物为吸附剂固相萃取分离盾叶薯蓣中的薯蓣皂苷时,在洗脱步骤中用5×1 mL H2O-C2H5OH(1∶1)混合液和5×1 mL H2O-CH3OH(1∶1)混合液洗脱时,可以回收86.01%的目标化合物,薯蓣皂苷得到了有效富集和分离,总回收率达99.66%。薯蓣皂苷经分子印迹萃取和分离后,产品中杂质大幅减少,薯蓣皂苷得到了较好的分离和纯化。另外,该种印迹聚合物可重复使用,吸附量改变不大。
猜你喜欢
基层中医药(2022年2期)2022-07-22
中成药(2018年10期)2018-10-26
天然产物研究与开发(2018年8期)2018-09-10
天然产物研究与开发(2018年5期)2018-06-13
中成药(2017年9期)2017-12-19
中国中医药信息杂志(2016年5期)2016-12-01
天然产物研究与开发(2016年1期)2016-06-05
医学研究杂志(2015年5期)2015-06-10
中国当代医药(2015年8期)2015-03-01
