
Solubility study of hydrogen in direct coal liquefaction solvent based on quantitative structure-property relationships model
2023-02-28 00:31XiaoBinZhangRajendranXingBaoWangWenYingLi
Xiao-Bin Zhang, A.Rajendran, Xing-Bao Wang,, Wen-Ying Li,
1 State Key Laboratory of Clean and Efficient Coal Utilization, Taiyuan University of Technology, Taiyuan 030024, China
2 College of Chemical Engineering and Technology, Taiyuan University of Technology, Taiyuan 030024, China
3 Department of Chemistry, Mepco Schlenk Engineering College (Autonomous), Sivakasi 626005, Tamil Nadu, India
Keywords:Hydrogen solubility Liquefied solvents Predictive model Density generalized function theory Quantitative structure-property relationship
ABSTRACT Direct coal liquefaction (DCL) is an important and effective method of converting coal into high-valueadded chemicals and fuel oil.In DCL,heating the direct coal liquefaction solvent(DCLS)from low to high temperature and pre-hydrogenation of the DCLS are critical steps.Therefore, studying the dissolution of hydrogen in DCLS under liquefaction conditions gains importance.However, it is difficult to precisely determine hydrogen solubility only by experiments, especially under the actual DCL conditions.To address this issue, we developed a prediction model of hydrogen solubility in a single solvent based on the machine-learning quantitative structure-property relationship (ML-QSPR) methods.The results showed that the squared correlation coefficient R2=0.92 and root mean square error RMSE=0.095,indicating the model’s good statistical performance.The external validation of the model also reveals excellent accuracy and predictive ability.Molecular polarization(α)is the main factor affecting the dissolution of hydrogen in DCLS.The hydrogen solubility in acyclic alkanes increases with increasing carbon number.Whereas in polycyclic aromatics,it decreases with increasing ring number,and in hydrogenated aromatics,it increases with hydrogenation degree.This work provides a new reference for the selection and proportioning of DCLS,i.e.,a solvent with higher hydrogen solubility can be added to provide active hydrogen for the reaction and thus reduce the hydrogen pressure.Besides,it brings important insight into the theoretical significance and practical value of the DCL.
1.Introduction
Direct coal liquefaction (DCL) has recently gained momentum as a clean and efficient coal utilization technology with low energy consumption[1,2].It converts coal with a H/C of ~0.8 into a hydrocarbon liquid fuel with a H/C of ~2.0,(H/C refers to the molar ratio of hydrogen and carbon elements in coal or solvent oil).In DCL,the catalyst and the oil slurry are mixed and fed into the hydrogenation reactor, where they react with the active hydrogen (H·) supplied by the hydrogen (H2) and direct coal liquefaction solvent(DCLS)at high temperature and pressure[3,4].During the liquefaction,the DCLS,besides being used to disperse and swell the pulverized coal particles, acts as a medium for heat transfer.Very importantly,the solvent dissolves hydrogen and improves the generation of active hydrogen to stabilize coal radical fragments and inhibit coking [5,6], as shown in Fig.1.
Yan et al.[7]investigated the solubility of hydrogen in tetrahydronaphthalene by the equilibrium sampling method.Hydrogen solubility in tetrahydronaphthalene increased with temperature and hydrogen partial pressure.Similarly, Fahim et al.[8] investigated the solubility of hydrogen in naphtha reformate cut that contains the components of coal-liquefied oil.Hydrogen solubility was greater in alkanes than that in aromatics.Notably, it decreased with the number of condensed aromatic rings in aromatics.Zhu et al.[9]determined the solubility of hydrogen in Shenhua coal liquefied oil at various temperatures (298.15-623.15 K) and pressures (2-10 MPa) using the equilibrium liquid phase sampling method.Like the abovementioned two reports,hydrogen solubility increased almost linearly with temperature and hydrogen pressure,emphasizing that hydrogen solubility increases with temperature and pressure regardless of the solvents.
Essentially,the hydrogen dissolved in the solvent participates in the hydrogenation reaction of the feedstock [10] and thus the quantity of dissolved hydrogen becomes a key factor in hydrogenation.As a consequence, researchers have recently determined the hydrogen solubility in a few solvents and heavy oils by applying experimental methods, including the equilibrium sampling method [11] and static analysis [12].Hydrogen first transfers to the liquid phase from the gas phase during the hydrogenation through physical dissolution [13].It enables the stabilization of free radical fragments of coal cracking via the improved interaction with the catalyst or solvent to yield the reactive hydrogen.Nevertheless, the results on hydrogen solubility and its impacts during DCL are limited due to the harsh reaction conditions(high temperature and pressure), the molecular complexity of DCLS, and the higher cost of a few solvents.Moreover, the solubility values obtained by experiments are often accompanied by significant uncertainties,leading to some difficulties in obtaining accurate dissolution patterns of hydrogen in solvents.Hence,it is of great theoretical interest and practical importance to establish a prediction model of solubility that relates hydrogen solubility and solvent structure.
Solubility is a quantitative indicator of the amount of hydrogen dissolved in a solvent.The quantum chemical molecular structure descriptors are reliable to obtain hydrogen solubility.As shown in Fig.2, we have developed a prediction model by the strategy employing the quantitative structure-property conformational relationship (QSPR) [14].Based on the quantitative relationship between the molecular structure of a compound and its physicochemical properties and solubility,we have established a quantitative relationship between the molecular structure of a solvent and hydrogen solubility.The density functional theory (DFT) method provides a better fit and prediction for the QSPR model than the semi-empirical AM1 algorithm [15].Selecting multiple molecular structure descriptors would reflect the molecular structure characteristics better and lead to multicollinearity among the independent variables.Cronauer et al.[16] found that the multiple linear regression (MLR) method can provide a better understanding of the properties of compounds via multiple molecular descriptors and relationships that offer useful insights into the factors affecting solubility.So far, the MLR method has been commonly used for developing solubility prediction models.
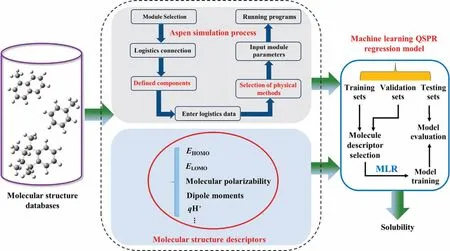
Fig.2.Solubility prediction model building flow chart.
Collecting sufficient data through experiments is difficult due to the uncertainty about the required hydrogen content under industrial conditions and the unavailability of solvent compounds in the market.Aspen simulation usually requires many physical parameters,which are hard to obtain for a few compounds of liquified oil.Thus, it becomes significant to represent the specific compounds by quantum chemical molecular structure descriptors to achieve comprehensive and in-depth research.Interestingly,this approach would be more accurate and intuitive to reflect the relationship between molecular structure and hydrogen solubility and more convenient for screening a solvent faster.
To the best of our knowledge,this is the first work to extend the ML-QSPR model to predict the solubility of gases in liquefied solvents.In this work, the solvent compounds have been selected based on the results of circulating solvent analysis in the Shenhua Ordos industrial plant [17,18].The QSPR prediction model of hydrogen solubility in a solvent has been established by combining quantum chemical molecular structure descriptors.An in-depth study on hydrogen solubility has been made from the perspective of the solvent molecular level structure.Also, all molecular and atom features for modeling have been derived from the molecular database to enable the construction of structure-property relationships.The relationship between the molecular structure of solvent and hydrogen solubility has been discussed from the perspectives of the number of branched chains, the number of aromatic rings,and the degree of hydrogenation.Our results have provided an important reference to select a suitable solvent at the correct proportion for the effective DCL.
2.Methods
In response to the current difficulties and the insufficient parameters of the properties of solvent compounds,a new method for solubility prediction was therefore established.By obtaining the optimal method for Aspen simulations and thus building the data set required for modeling, quantum chemical calculations were used to obtain the molecular structure descriptors for each solvent compound.Then the data set was classified and the MLQSPR model was built using the training set, and the reliability of the model was evaluated using the test and validation sets.
2.1.Database preparation and partitioning
Since our model aims to accurately predict the hydrogen solubility in liquefaction solvents, a reliable dataset becomes essential to develop the prediction model[19].The solvent compounds were mainly derived from the Shenhua DCL unit,which mostly includes compounds such as acyclic alkanes, monocyclic aromatics, and polycyclic aromatics.The aromatics hydrocarbons in them contain partially hydrogenated aromatic hydrocarbons and fully hydrogenated aromatic hydrocarbons.Due to the harsh experimental conditions associated with gas-liquid equilibrium, this paper adopts the powerful database of Aspen to obtain the hydrogen solubility in single-component solvents to build a dataset.It reflects real liquefaction reaction temperature and pressure.The sudden pressure drop in the gas-liquid equilibrium flashes out the solute gas from the solvent to achieve gas-liquid separation, ensuring the smooth running of Aspen simulations.Therefore, it is critical to identify an appropriate two-phase feed rate(gas-liquid)to guarantee the achievement of full saturation while mixing gas and liquid [20].
Fig.3 shows that the change of the solvent flow rate below 1000 kg·s-1does not have any effect on the dissolved hydrogen concentration, implying complete saturation.The hydrogen concentration in the liquid increases until the liquid reaches saturation at the hydrogen flow rate(2 kg·s-1)due to the increase in the ratio of hydrogen to solvent flow rate.Hence,we conclude that the ratio of hydrogen to solvent flow rate needs to be greater than 1:500 in the simulated hydrogen dissolution equilibrium.
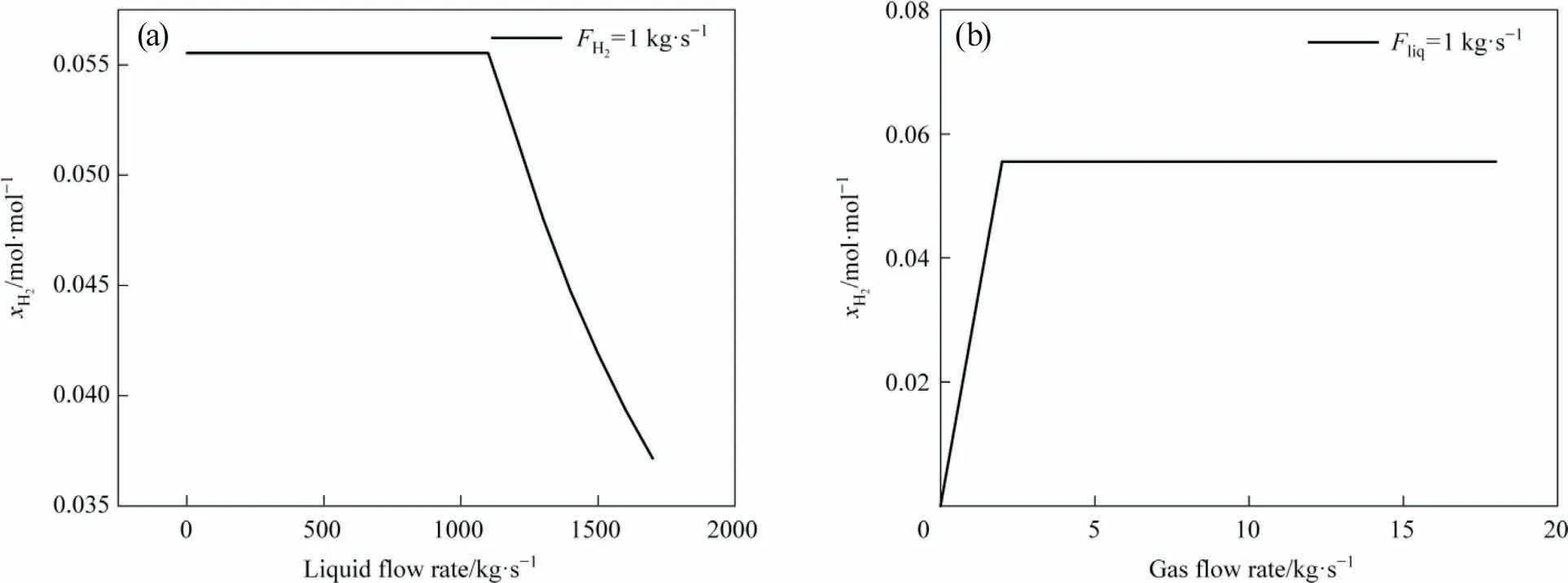
Fig.3.Determination of hydrogen and solvent flow rates:(a)fixed hydrogen flow rate and variable solvent flow rate;(b)fixed solvent flow rate and variable hydrogen flow rate).
As shown in Fig.4,Hydrogen solubility in hexadecane, toluene,naphthalene, and phenanthrene calculated via the different simulation methods has been compared with the experimental results[21-23].Amidst them, the solubility values calculated using SRK (Soave-Redlich-Kwong) are very close to the corresponding experimental data, SRK is a thermodynamic equation of state.Therefore, the SRK method has been employed in the subsequent simulations.
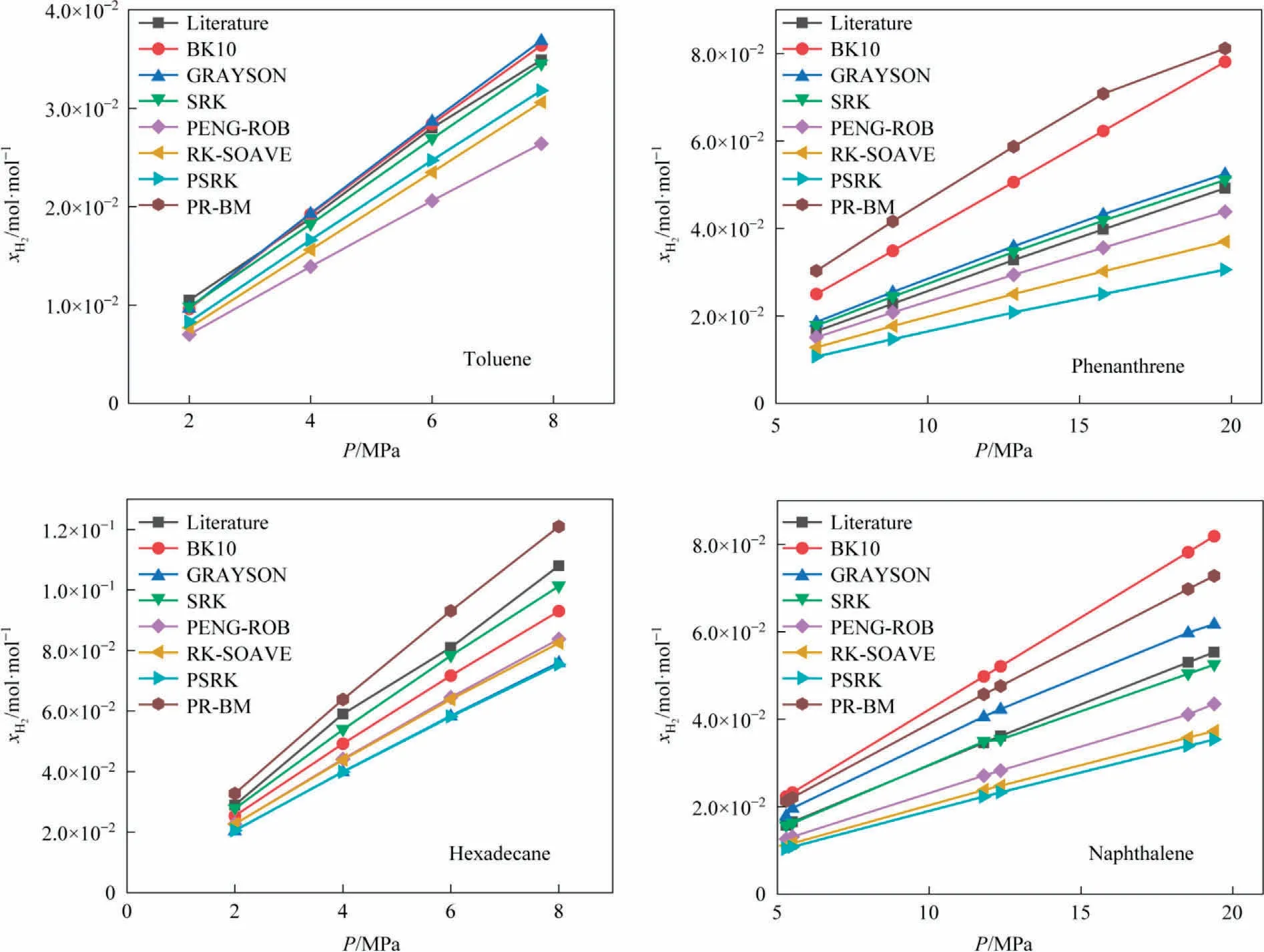
Fig.4.Comparison of simulated and experimental values of hydrogen solubility in hexadecane, toluene, naphthalene, and phenanthrene (T = 180 °C).
The simulations have been performed by following the industrial DCL conditions (T = 723 K and P = 18.5 MPa) [24].The calculated solubility values (molar fraction (X)) were calculated as X =nGas/ (nGas+nSolvent) and are in the range of 0.160-0.296.The lnX values were calculated to attain a reliable model, which does not alter the nature of the data.Beneficially,it compresses the scale of the variables and may lead to obtaining smooth data between the descriptors of the model [25].
2.2.Descriptor calculation
A molecular structure descriptor is a measure of a molecule’s properties in a particular aspect.It can be a physicochemical property of the molecule, or an algorithmically calculated numerical index [26,27].The adopted molecular structure descriptors facilitate the interpretation of the model mechanism.The molecular structure descriptors exhibiting physicochemical properties are preferable[28].The molecular structure descriptors were comprehensively calculated using the Gaussian 16 package [29].Firstly,the molecular structure of the compound was optimized based on the general function combined with the 6-31+G(d,p) basis group.Frequency calculations were carried out to guarantee the lowest energy on the potential energy surface [30,31].In light of the optimal structure,the single-point energy calculation was done considering the 6-31+G(2df,2p)basis group[32].Likewise,we have calculated the molecular descriptors,and the calculated values are given in Tables S1-S3 in Supplementary Material.
2.3.Descriptor selection
The decrease in the accuracy of the computation often arises due to irrelevance in the modeling of structure-property relationships.Hence,screening molecular descriptors is important to build a robust and accurate prediction model without over-correlation.The descriptors were ranked based on the magnitude of the correlation coefficient relating the solubility with them.The molecular descriptors are filtered using the feature selection method of random forest.This method considers forward selection to determine the optimal subset of features with the highest modeling accuracy until there are no left features.Through this filtering,six molecular structure descriptors have been comparatively screened, which shows a high correlation with hydrogen solubility.These descriptors are molecular polarization(α),dipole moment(μ),the highest occupied orbital energy (EHOMO), the lowest unoccupied empty orbital energy (ELUMO), the most positive net charge of hydrogen atom(qH+), and the most negative charge of the carbon atom(q-).
2.4.Modeling architecture
The QSPR study of hydrogen solubility in solvents was carried out based on the traditional MLR and modern machine learning(support vector machines) approaches.Relying on solid statistical theory, MLR can cope with a variety of experimental datasets.Recent years have witnessed many new linear modeling methods as reliable alternatives to traditional linear statistics, which are often appropriate for solving regression and classification problems.
The balanced subset method was utilized for the dataset partition that constructs a reliable model and ensures that the structure-property relationships are similar [33].It divides the complete dataset into three subsets, i.e., the training set (35 compounds) used for model development.The hyperparameters were manually tuned and optimized using a trial-and-error method using algorithm engineers.The training data was divided into two individual subsets.One subset was used to learn the parameters and the other was used as a validation set to estimate the generalization error during or after training, and thus the hyperparameters were updated.The training set was built by employing the statistical molecular design[34]to reveal statistical randomness, structural representativeness, and comprehensiveness.Also, a few basic conditions (e.g., the reliability of the nature data) were satisfied well.In addition, a validation set (25 compounds)and a testing set(10 compounds)were developed for testing the model and true external validation,respectively.The model was described using the following equation.
where Y is the dependent variable(solubility);X1···Xmare the independent variables (molecular descriptors); b0is the intercept (constant term); b1···bmare the regression coefficients.
The independent variables are often assumed to be orthogonal in the MLR process.Here, a predictive model was created based on the modified theory of linear solvation energy relations(MTLSER) and mechanistically analyzed at the microscopic level.Samuel et al.[35] used MTLSER to investigate the correlation between different solvent parameters.It showed the existence of a dipole moment/molecular polarization effect between solute and solvent molecules.Famini et al.[36] and Toropov et al.[37]replaced the solvation parameter with a quantitative parameter in the linear solvation energy relationship to propose a theoretical linear solvation energy relationship.Based on this theory, Chen et al.[38-40] further refined the modified linear solvation energy relationship model by relating three processes associated with free energy.A cavity to accommodate solute molecules was first formed in the solvent molecules (endothermic) followed by the separation of solute molecules that enter the cavity (exothermic),leading to the solute-solvent interaction.This is the primary concept reflecting the dissolution of hydrogen in the solvent.
2.5.Modeling evaluation
2.5.1.Fitting
Multivariate calibration methods are commonly used for generating QSPR models, including MLR analysis, partial least squares regression analysis, nonlinear regression analysis, artificial neural network methods,and MLR models[41].For the same,a free software package (IBM SPSS 12.0) was used in this work [42].
The coefficient of determination (R2) (Eq.(2) is often used as a key indicator to ensure the goodness of fit in the model.The larger the R2value, the better the goodness of fit is.The calculation formula is given below.
2.5.2.Modeling reliability validation
The stability and predictivity of the model were characterized using a simulated external validation method.Several solvent compounds were chosen as the testing set,which was divided into a training set and a validation set.The training set is used to build up the model and the validation set is used to check the reliability of the model.The predictive ability of the model was reflected by the resulting Q2,which was compared with the R2values,reflecting the statistical stability.Moreover, the variance inflation factors(VIF) were applied to assess the correlation degree of the respective variables.If VIF <5, there was no multicollinearity between the independent variables.The residual distribution effectively revealed the stability of the model.
3.Results and Discussion
This paper discusses how hydrogen solubility is influenced by temperature and pressure on the hydrogen solubility and molecular structures of various solvents.In this context,the six molecular structure descriptors were obtained from the screening calculation as independent variables.A stepwise MLR analysis was carried out on the hydrogen solubility values in the training set to establish a model predicting the hydrogen solubility in the chosen solvents.The validation test was applied to choose the optimal model for predicting hydrogen solubility.Tables S1-S3 reveal the hydrogen solubility values calculated from the Aspen simulations.
3.1.Effect of process parameters
3.1.1.Temperature
The effect of temperature on hydrogen dissolution in tetrahydronaphthalene at different temperatures (Fig.5).Hydrogen solubility in tetrahydronaphthalene has a linear relationship with temperature.Henry’s law applies to the dissolution of insoluble gases under low-pressure conditions but the system is under high-pressure conditions.The increase in temperature improves the molecular movement of the gas and increases the pressure,resulting in enhanced hydrogen dissolution.Nonetheless, a very high temperature is not preferable as it promotes coke generation[39-41].The optimal temperature for the Shenhua DCL is ~723 K and increasing the temperature beyond a certain limit might not have a significant effect as evidenced by Fig.5.Therefore, a dynamic equilibrium process is maintained, i.e., hydrogen content in the solvent is in a dynamic equilibrium process of depletion and replenishment.
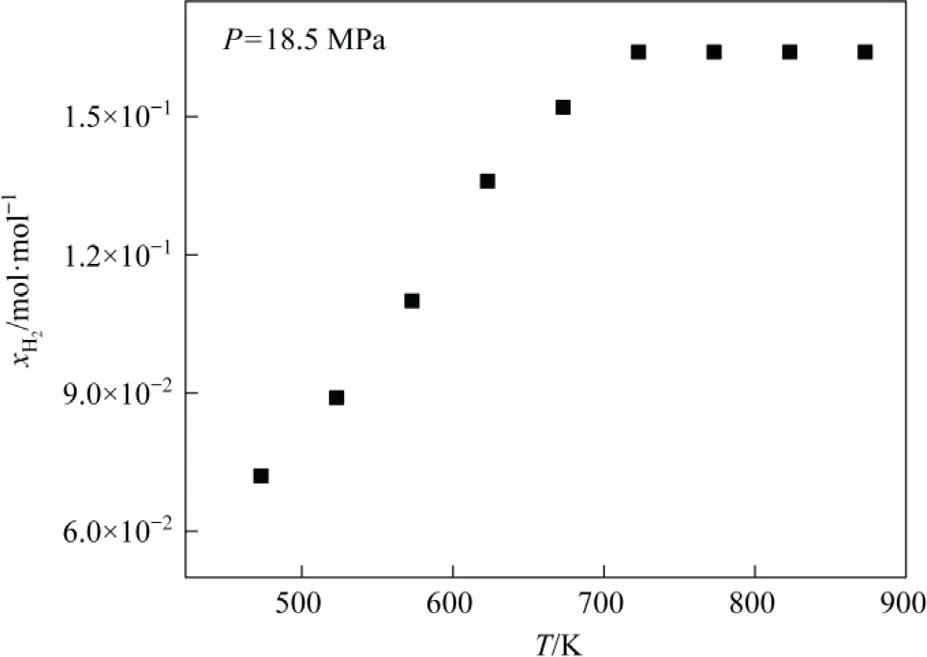
Fig.5.Hydrogen solubility in tetrahydronaphthalene at different temperatures.
3.1.2.Pressure
Fig.6 illustrates the impact of pressure on the hydrogen solubility in tetrahydronaphthalene.Hydrogen solubility increases with pressure, which underlines the importance of maintaining high pressure in the DCL [43-45].However, from the perspectives of economy and safety, a moderate hydrogen pressure is advisable in the DCL, which is currently limited to less than 20 MPa.As shown in Fig.6, the hydrogen solubility in tetrahydronaphthalene undergoes a negligible change at pressures higher than 13 MPa.
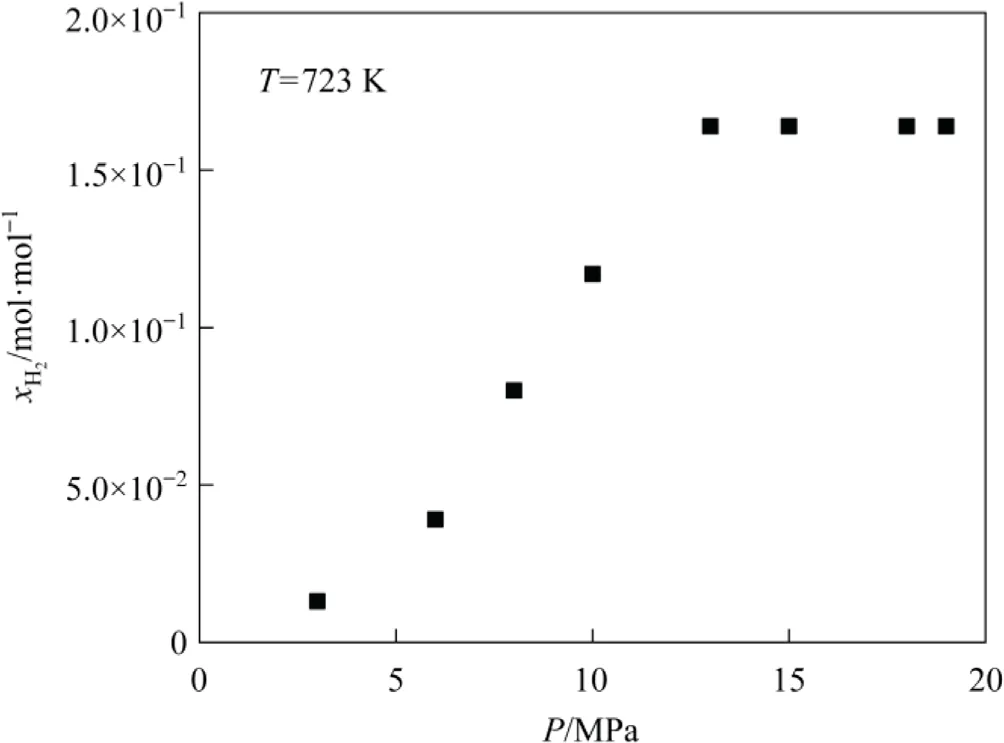
Fig.6.Hydrogen solubility in tetrahydronaphthalene at different pressures.
3.2.Modelling results
The structural parameters were obtained from the quantitative calculations and applied as theoretical descriptors.The correlation equation between solubility and theoretical descriptors was established using the GQSARF 2.0 program.The optimal model derived by the stepwise regression method is given below.
Then she commanded her servants to cast Death out of the city, which they did, with such hard blows that he never dared to show his face again in the Land of Immortality
The best descriptors needed to build the model were identified using the stepwise regression method.Different numbers of molecular descriptors were introduced in the prediction model.The introduction of five molecular structure descriptor parameters attains the maximum coefficient of determination (R2= 0.917 >0.9)(Table S4),implying that the model has a good fit and the equation is more relevant.The results of the six models obtained after screening are given in Table S4.The scenarios involving 1 to 6 molecular structure descriptors were investigated.Though the models containing 4 and 5 molecular structure descriptors are similar, the difference between RMSEtrainand RMSEvalis relatively small for the models containing 5 descriptors.As a result, the solubility prediction model with 5 descriptors was developed.
Table S5 indicates that the constants,α, ELUMO,and qH+have a certain correlation with hydrogen solubility.Comparing the significance of each descriptor of the built model suggests a positive influence of descriptors on hydrogen solubility though their degree of influence is not the same.In line with the normalized coefficient values of the descriptors,the molecular polarizability has a greater effect than the lowest vacancy orbital energy(ELUMO)and the most positive net charge in the hydrogen ion(qH+).The variance expansion factor is less than 5 (VIF <5) and shows no multicollinearity among the molecular descriptors.
The prediction results of the prediction model were obtained with the ML-QSPR method(Fig.7).The R2connecting the predicted and measured values is 0.92.The normalized residuals of the predicted values (Fig.8) suggest the stability of the developed model.The residuals are defined as the differences between the simulation and predicted values and about 88% of residuals are within the error range.
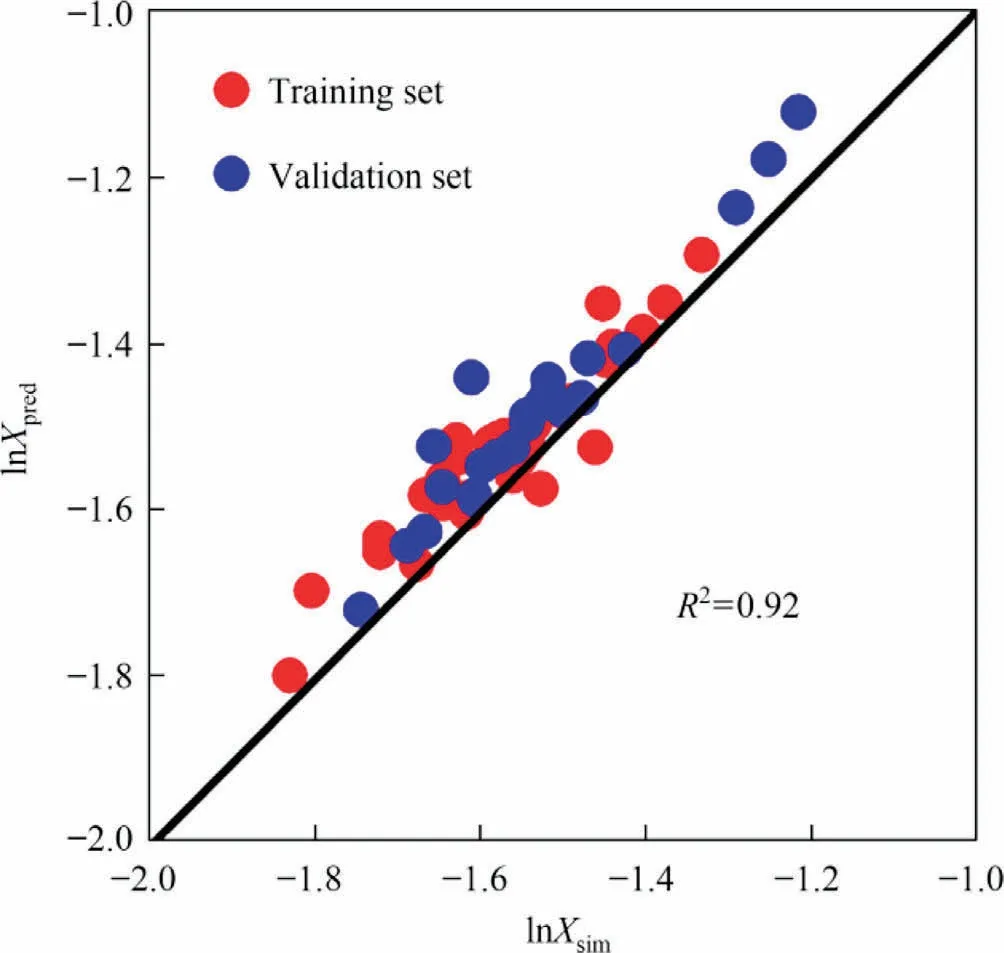
Fig.7.Fitted relationship between simulated and predicted solubility values.
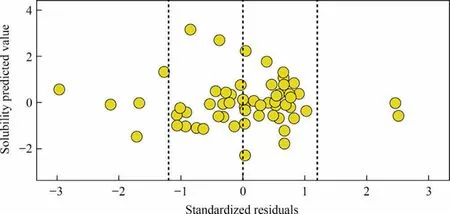
Fig.8.Fitted relationship between simulated and predicted solubility values.
The obtained results highlight the good stability and prediction capacity of the prediction model.In the modeling, the ratio of the number of samples to the number of descriptors was considered greater than 4:1 to prevent false regression due to the chance of correlation among too many descriptors.
The maximum relative deviations of the QSPR models are below 10% and the average absolute relative deviation (AARD) distribution of the entire dataset is shown in Fig.9.20% of the data points in the whole dataset are within 0%-1%deviation,49.23%are within 1%-5%, and 30.77% are within 5%-10%.
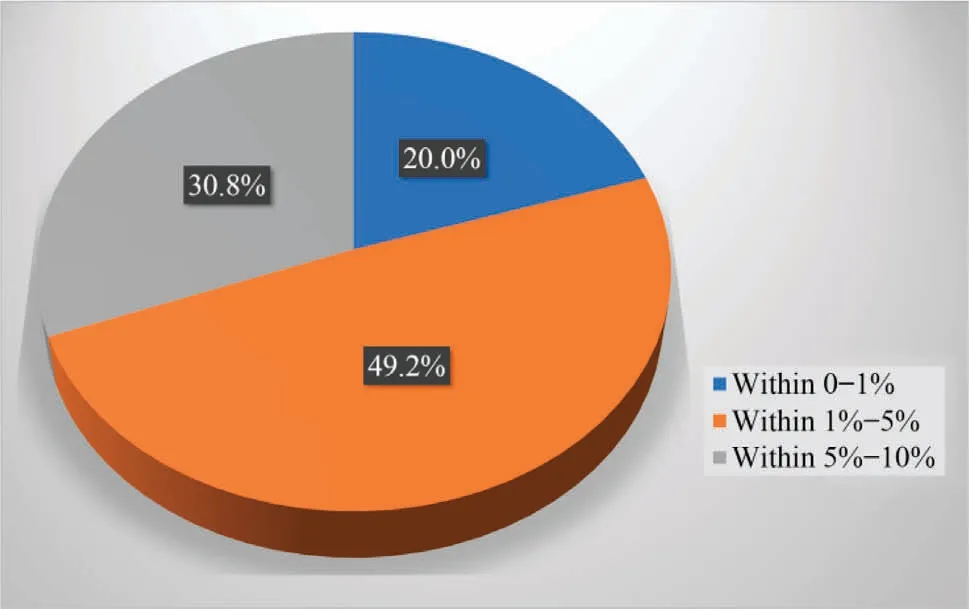
Fig.9.Percent values within different deviations of the QSPR model.20%, 49.2%,and 30.8% of the H2 solubility in the solvent are within 0%-1%,1%-5%,and 5%-10%(relative deviation range), respectively.
3.3.Mechanistic analysis of model
The model was constructed by analyzing the mechanism from the perspective of the molecular structure descriptors.It contains a total of three molecular structure descriptors, namely, α, ELUMO,and qH+.Here, α has the strongest correlation to lnX with the R2value of 0.903 (a positive coefficient), underlining that molecular polarization rate (α) has a significant influence on the hydrogen solubility in the solvent.The solubility gradually increases with the molecular polarizability due to the linear correlation between the α values and the molecular volume.As a result, the higher molecular volume, i.e., the higher α value, leads to a larger gap between the solvent molecules, and thus less energy is required for hydrogen molecules to enter the solvent.The hydrogen solubility increases with ELUMO, revealing a charge transfer interaction between hydrogen and solvent molecules.qH+indicates the ability of molecules to provide protons or accept electrons, which has a positive coefficient in the equation.Moreover, it is closely related to the formation of intermolecular H-bonds and the polar interaction of molecules.Therefore, with the increase of qH+, hydrogen may easily enter into the solvent molecules of higher polarity,which is positively correlated with lnX.
3.4.Model validation
VIF was used to evaluate the degree of correlation between the respective variables in the equation and calculated from the equation VIF = 1/(1-r2) (r is the correlation coefficient of the multiple regression between one independent variable and the other variables).If VIF = 1.0, no autocorrelation exists among the variables.On the other hand, the VIF between 1.0 and 5.0 shows that each independent variable of the correlation equation is uncorrelated.If VIF >10, the regression equation is unstable and requires a retest.The VIF values among the six independent variables in our model equation are below 5,suggesting the absence of correlation between the independent variables in the model.The Debin-Watson values are close to 2 and indicate the independent nature of the selected samples.The cross-validation coefficient is 0.908(Table S6), which underpins the good stability and predictive ability of the established model.
3.5.Hydrogen solubility in various solvents
Hydrogen solubility was comparatively investigated in acyclic alkanes and aromatics under the DCL conditions (450 °C and 18.5 MPa).It reveals the relationship between hydrogen solubility and the molecular structure of the chosen solvent from a microscopic perspective.The obtained results are shown in Fig.10.The hydrogen solubility is higher in the acyclic alkanes and very less in the tetracyclic aromatics.
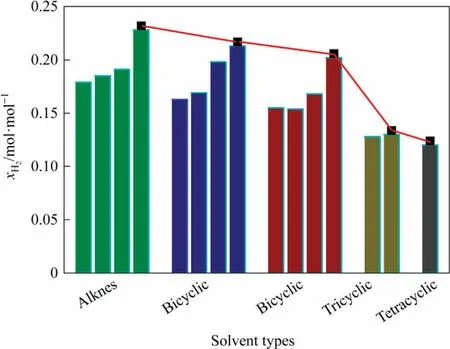
Fig.10.Comparison of the hydrogen solubility values in different families of liquefied solvents at 450 °C and 18.5 MPa.
3.5.1.Hydrogen solubility in different acyclic alkanes
The hydrogen solubility increases with the number of carbon atoms present in acyclic alkanes.The molecular volume enlarges with the chain length of acyclic alkanes, resulting in the gap enhancement between the solvent molecules.Therefore, the hydrogen solubility in acyclic alkanes is mostly influenced by the polarization rate of molecules, leading to a gradual increase in hydrogen solubility.The branched alkanes exhibit a higher molecular polarization rate than the straight-chain alkanes.Hence, the branched alkanes could dissolve hydrogen better than the straight-chain alkanes containing the same carbon number.
3.5.2.Hydrogen solubility in aromatics with different ring numbers
In comparison,acyclic alkanes are better solvents than aromatics to dissolve hydrogen molecules.In the case of aromatics,the hydrogen solubility gradually decreases with the number of aromatic rings of the concerned aromatic solvent.Concerning the number of aromatic rings, the efficiency order for the aromatic solvents to dissolve hydrogen is monocyclic >bicyclic >tricyclic >tetracyclic.It is because the increase in the number of aromatic rings promotes the interaction between macromolecules.As a result, hydrogen molecules could not easily enter the solvent molecules with lower energy.
3.6.Hydrogen solubility in hydrogenated aromatics
The liquefaction reaction can proceed through two different routes.Hence,the hydrogenated aromatics partially hydrogenated in circulating solvents show different solubility of hydrogen in them.In the thermal route,the active hydrogen for the liquefaction is mainly provided by the solvent [7,41].Differently, in the catalytic route,the catalyst activates the hydrogen molecule to supply active hydrogen for liquefaction.Bai et al.[9,46] and Wang et al.[47] explored the hydrogen supply activity of different hydrogenated aromatics during the liquefaction and proved that the active hydrogen is mainly provided by the solvent during the thermal liquefaction.Fig.11 shows the variation of hydrogen solubility in different hydrogenated aromatic solvents.
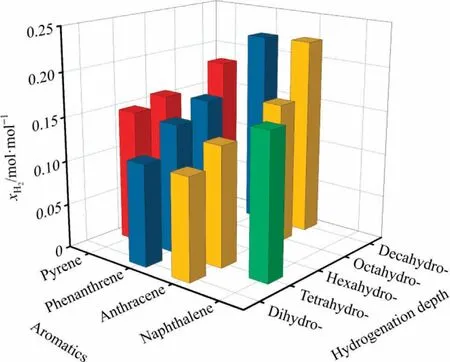
Fig.11.Hydrogen solubility in different hydrogenated aromatics.
3.6.1.Hydrogen solubility in tetrahydronaphthalene with different substituents
In tetrahydronaphthalene containing different substituents,the hydrogen solubility varies with the number of substituents.The higher the number of substituents in tetrahydronaphthalene, the greater the hydrogen solubility is.It is due to the gradual increase of the polarization rate of solvent molecules with the number of substituents.The resultant increase in the molecular volume boosts the voids between solvent molecules and makes it easier for hydrogen molecules to enter the voids, leading to the effective dissolution of hydrogen molecules in the solvent.
3.6.2.Hydrogen solubility in hydrogenated anthracenes,hydrogenated phenanthrenes, and hydrogenated pyrenes
The hydrogen solubility varies with the hydrogenation degree of the hydrogenated anthracene molecules, which are partially hydrogenated in the liquefied solvent.The hydrogen solubility increases with the hydrogenation degree of hydrogenated aromatics because the increase in hydrogenation degree increases the polarization rate of solvent molecules.However, the quantity ELUMOgradually increases,implying that the ability to accept electrons gradually decreases and the molecular motion becomes more intense.Consequently, the hydrogen solubility in the solvent increases with ELUMO.Note that the hydrogen solubility in 9,10-dihydroanthracene is smaller than that in other hydrogenated anthracenes.In the literature, the H-donating capacity of 9,10-dihydroanthracene during liquefaction is greater than that of other hydrogenated anthracenes[9,46,48].In this view,the active hydrogen in this process mainly generates from the solvent rather than hydrogen.It also supports that the solubility of hydrogen in 9,10-dihydroanthracene is not effective.Similar observations are also noticed for the hydrogen solubility in hydrogenated phenanthrenes and hydrogenated pyrenes.
4.Conclusions
In this paper, we calculated the quantum chemical descriptors of a single solvent molecule in liquefied solvents by the density generalized functional theory method and constructed a QSPR model for predicting the solubility of hydrogen.The uniqueness of this work is that it replenished the solubility studies from a perspective of molecular level.The models with different groups of descriptors have proven that the QSPR model has a good statistical performance.Moreover,the validation of the model implies that it has a good predictive ability.The simulated and predicted values exhibit a good agreement.Based upon forward selection involving the Random Forest method, the five quantum chemical molecular structure descriptors have been applied.
The hydrogen solubility values in various solvent compounds have been studied using the tested model.The predicted values correlate strongly with the simulated values showing R2and RMSE as 0.92 and 0.095, respectively.
The final models were validated to be statistically reliable and predictive using the leave-one-out cross-validation and an external test set.The comparison with reference models demonstrates that this new method is very efficient and provides satisfactory results in significant improvements,both in accuracy and stability for predicting the hydrogen solubility in a particular solvent.Acyclic alkanes, aromatics, and hydrogenated aromatics were the studied solvents due to their presence in liquefied solvents.The number of carbons in acyclic alkane solvents, the number of rings in the aromatic solvents, and the hydrogenation degree in the hydrogenated aromatics are the significant factors that influence hydrogen solubility.The polarization rate (α) linked with the molecular volume of the solvents plays a dominant role in determining hydrogen solubility.Briefly,this work provided a modeling method for predicting hydrogen solubility in pure solvents.Importantly,this method can be extended with the generation of more descriptors to predict the physicochemical properties of chemical compounds.
Data Availability
No data was used for the research described in the article.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors are very grateful for the financial support from the National Key Research and Development Program of China(2022YFB4101302-01), the National Natural Science Foundation of China (22178243) and the science and technology innovation project of China Shenhua Coal to Liquid and Chemical Company Limited (MZYHG-22-02).
Supplementary Material
Supplementary material to this article can be found online at https://doi.org/10.1016/j.cjche.2023.05.014.
 Chinese Journal of Chemical Engineering2023年12期
Chinese Journal of Chemical Engineering2023年12期
- Chinese Journal of Chemical Engineering的其它文章
- Intrinsic kinetics of catalytic hydrogenation of 2-nitro-4-acetylamino anisole to 2-amino-4-acetylamino anisole over Raney nickel catalyst
- Experiments and model development of p-nitrochlorobenzene and naphthalene purification in a continuous tower melting crystallizer
- α-Synuclein: A fusion chaperone significantly boosting the enzymatic performance of PET hydrolase
- Influence of water vapor on the separation of volatile organic compound/nitrogen mixture by polydimethylsiloxane membrane
- Mass transfer mechanism and relationship of gas-liquid annular flow in a microfluidic cross-junction device
- Enhanced photocatalytic activity of methylene blue using heterojunction Ag@TiO2 nanocomposite: Mechanistic and optimization study