
α-Synuclein: A fusion chaperone significantly boosting the enzymatic performance of PET hydrolase
2023-02-28 00:30RenwenTianYanSun
Renwen Tian, Yan Sun
Department of Biochemical Engineering, School of Chemical Engineering and Technology and Key Laboratory of Systems Bioengineering and Frontiers Science Center for Synthetic Biology (Ministry of Education), Tianjin University, Tianjin 300350, China
Keywords:PET hydrolase α-Synuclein Fusion enzyme Stability Pollution Degradation
ABSTRACT Extensive use of polyethylene terephthalate (PET) has brought about global environmental problems.A recently reported PET hydrolase (PETase) discovered from Ideonella sakaiensis showed high potential for degrading PET at moderate temperatures, but its activity and stability need further improvement for practical applications.Herein,we proposed to use α-synuclein(αS)as a fusion chaperone and created six PETase-αS fusion enzymes with linkers of different types and lengths.All the fusion enzymes exhibited improved enzymatic performance, presenting 1.5 to 2.6-fold higher activity towards bis-2(hydroxyethyl) terephthalate than PETase, as well as significantly increased stabilities.Fluorescence spectroscopy indicated that the chaperone fusion tightened the overall conformation and resulted in the opening of the substrate binding pocket, which led to the improved thermal stability and catalytic activity of the fusion enzymes.Remarkably, one of the fusion proteins, PETase-[(GS)(EK)]10-αS, showed 3.2 to 5.1 times higher PET degradation capability than PETase.The significantly boosted PET degradation performance was not only attributed to the enhanced enzymatic activity and stability, but also possibly due to the binding affinity of the fused αS domain for PET.These findings demonstrated that αS was an effective fusion chaperone for significantly enhancing the enzymatic performance of PETase.
1.Introduction
Plastic is an essential material in modern society,and polyethylene terephthalate (PET) currently stands as the most abundantly manufactured polyester plastic [1].PET is an aromatic polyester,composed of terephthalic acid (TPA) and ethylene glycol (EG),which polymerize via ester bonds,conferring PET with exceptional properties such as strength, transparency, durability, and nontoxicity [2,3].PET’s worldwide production surpasses an estimated 70 million tons per annum [4].Despite its numerous benefits, a considerable amount of PET waste arises from discarded PET packaging materials after use, leading to a substantial burden on the environment [5].This phenomenon is of great concern, given the magnitude of PET waste that enters landfills and oceans [6].
The disposal of polyethylene terephthalate (PET) waste commonly involves landfill,incineration,and recycling[7],all of which can generate harmful byproducts and lead to secondary pollution.As an alternative,enzymatic degradation has emerged as a promising and sustainable approach that has received extensive research attention.Lipase, esterase, and keratinase are among the enzymes that have demonstrated PET hydrolysis activity[8,9].Nevertheless,these enzymes typically exhibit optimal hydrolysis temperature around the glass transition temperature of PET (Tg= 65 °C) [10].In 2016, Yoshida et al.[11] discovered Ideonella sakaiensis 201-F6,a novel bacterium that utilizes PET as its primary carbon source and secretes PET hydrolase (PETase), an enzyme capable of hydrolyzing PET into smaller molecules.PETase has demonstrated 5.5-120 fold higher PET degradation activity than the abovementioned enzymes at a moderate temperature (30 °C) [11,12],making it a more promising candidate for PET biodegradation.Through exploration of the crystal structure of PETase[12],the catalytic mechanism has been elucidated.The rational protein engineering has resulted in the generation of a diverse PETase mutants that exhibited improved hydrolytic activity and/or thermal stability [13-16].For example, rational protein engineering has also led to the creation of PETaseS121E/D186H/R280Avariants with 14-fold higher PET degradation activity than native PETase [15].Numerous studies have been dedicated to enhancing the catalytic efficiency of PETase in the biodegradation of PET [17-20].However,the activity and stability of PETase remain critical factors that limit its industrial applications.
Enzyme modification and redesign through molecular approaches have become increasingly important for overcoming the limitations of enzyme-catalyzed industrial applications.As an effective addition to protein modification and redesign, genetic fusion strategy based on fusion protein design has emerged as an effective method for enhancing enzyme performance via various molecular strategies [21].The most widely used strategy is endto-end fusion, which involves linking another protein/enzyme or peptide directly or through a linker peptide to one or both of the target enzyme termini.A proper linker can function to separate the two parts,thereby preventing mutual interference during folding and catalysis[22],and improving the activity and/or stability of the target enzyme [23-25].
α-Synuclein (αS) is a 140-residue intrinsically disordered protein that is highly expressed in presynaptic terminals and implicated in the pathogenesis of Parkinson’s disease [26].This protein comprises three distinct domains, namely the basic Nterminal domain (residues 1-60), the hydrophobic nonamyloid-β component (NAC, residues 61-95), and the acidic C-terminal domain (residues 96-140) [27].It was reported in 2020 that the co-incubation of alcohol dehydrogenase (ADH) with free αS could stabilize ADH, and gold nanoparticles bound with the 16-residue peptide (αSP) from the C-terminal sequence of αS could adhere to various substrates,including PET[28].Recently,our lab reported that the fusion of αSP with cutinase ICCG resulted in an increased degradation efficiency of PET [29].Therefore, we have herein proposed to use αS as a fusion chaperone to explore its stabilizing effect on PETase.Moreover, by attaching αS to the C-terminal of PETase to allow the C-terminal of αS free, its adhesive property would improve the enzyme affinity for PET, thus enhancing the enzymatic PET degradation performance of the fusion enzyme.To this purpose,six different fusion enzymes were designed with different peptide linkers and linker lengths.The enzymatic performance of the fusion enzymes was extensively studied and compared with the native enzyme to provide insight into the development of high-performance PET hydrolases.
2.Materials and Methods
2.1.Materials
Bis-2(hydroxyethyl) terephthalate (BHET), terephthalic acid(TPA) and fluoresceine isothiocyanate (FITC) were purchased from Aladdin (Shanghai, China).Mono(2-hydroxyethyl) terephthalate(MHET) was obtained from MOLBASE (Shanghai, China).Sodium dodecyl sulfate (SDS) was obtained from Solarbio (Beijing, China).Nongfu Mountain Spring bottle films were obtained from commercial sources (Tianjin Nongfu Shanquan Beverage, Tianjin, China).Escherichia coli (E.coli) BL21 (DE3) and Shuffle T7 competent cells were from Solarbio(Beijing,China).All the other chemical reagents used in this study were of analytical grade.
2.2.Enzyme preparation
Alpha-synuclein (αS, NCBI Reference Sequence: NP_000336.1)from Homo sapiens were fused to the C-terminus of PETase and connected by six different linkers (Table S1 in Supplementary Material), resulting in six fusion enzymes, named PETase-(GS)3-αS, PETase-(GS)6-αS, PETase-(GS)20-αS, PETase-(EK)20-αS, PETase-[(GS)(EK)]10-αS, and PETase-[(GS)(EK)]20-αS.The PETase gene(GenBank accession number: GAP38373.1) without signal peptide(amino acid residues 30-290) and the gene encoding for fusion enzymes were codon-optimized, synthesized by Azenta (Suzhou,China).The PETase gene segment was subcloned into the pET-22b (+) vector and the six fusion enzyme fragments were subcloned into pCold II vectors.Additionally, the resulting expression plasmid pET-22b (+): PETase was transformed into E.coli BL21(DE3), and plasmids pCold II: PETase-(GS)3-αS, pCold II: PETase-(GS)6-αS, pCold II: PETase-(GS)20-αS, pCold II: PETase-(EK)20-αS,pCold II: PETase-[(GS)(EK)]10-αS, and pCold II: PETase-[(GS)(EK)]20-αS were transformed into E.coli Shuffle T7.All the fusion enzymes carried an N-terminal 6 × His Tag for purification by immobilized metal affinity chromatography (IMAC) with 1-ml packed column of nitrilotriacetic acid chelated nickel (Ni-NTASepharose, GE Healthcare) column (Tricorn 5/50, 5 × 50 mm, GE Healthcare).The protein expression and purification were analyzed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE).
PETase and its fusion enzymes expressed in E.coli were cultured,recovered and purified as described following the procedure described by Chen et al.[30].The αS was expressed and purified following the procedure described previously [31].
2.3.Enzymatic performance assays with BHET
Enzyme activities of PETase and its fusion enzymes were measured using BHET as substrate at 25 - 50 °C as described previously [30].The enzyme activity was defined as the concentration(μmol·L-1)of MHET released by the enzyme(50 nmol·L-1)catalyzing the degradation of BHET(200 mg·L-1)for 30 min[15,32,33].All the experiments were performed in triplicate and the average values and standard deviations(SD)were reported.HPLC experiments for product analysis were also performed by the literature method[30] with a Welch Ultimate XB-C18 column (4.6 mm × 250 mm,5 μm, Welch Materials, Shanghai, China).The concentrations of various products were calculated from the standard curves(Fig.S1).
The thermal stabilities of PETase and its fusion enzymes were investigated by incubating the enzymes (5 μmol·L-1) at 40 °C for different time (0 - 6 h).After incubation, the enzyme solution was quickly cooled down on the ice, followed by activity measurement as described above at 35 °C.For comparison of residual activities, the initial activity prior to heat treatment was set as 100%.
The melting temperature (Tm) of the proteins were determined by established methods with minor modifications [34].In brief,thermal denaturation curves were obtained by a Jasco J-810 circular dichroism (CD) spectrometer (JASCO, Tokyo, Japan) by gradually increasing the temperature from 25 °C to 65°C at a heating rate of 2 °C·min-1at the wavelength range of 200-230 nm.The signal at 222 nm was recorded as a function of temperature, since the CD thermal scan in the far-UV CD region (222 nm) is known to reflect changes in the content of the α-helical conformation.Tmvalues were calculated by the Boltzmann curves fitting of the ellipticityversustemperature data.
The kinetics of the enzymatic reactions were determined by measuring the initial catalytic activity with BHET at 0.5 to 18 μmol·L-1at 35 °C.The initial rate was calculated using the initial linear hydrolysis and plotted against BHET concentrations.The kinetic data were analyzed based on the Michaelis-Menten kinetics described by the following equations [30,35],
where v is the reaction rate (mmol·L-1·min-1), [S] is BHET concentration (mmol·L-1), Vmaxis the maximum reaction rate(mmol·L-1·min-1), Kmis the Michaelis constant (mmol·L-1), [E0]represents the enzyme concentration in the reaction system(50 nmol·L-1), and kcatis the reaction constant (turnover number)(min-1).All experiments were performed in triplicate.
2.4.Spectroscopic measurements
Fluorescence analysis was carried out following Chen et al.[30].Fluorescence spectra of the enzymes were analyzed by Luminescence Spectrometer (PerkinElmer, USA).The shift in maximum wavelength of emission spectra was utilized to monitor the conformational changes in PETase induced by αS fusion.
2.5.Degradation of PET films
To analyze the degradation efficiency of PET films by PETase and its fusion enzymes, commercial PET film for drinking water packaging was used.The crystallinity of the Nongfu Mountain Spring bottle films was measured by the differential scanning calorimetry(DSC)as described earlier[4,14,15].Determination of film degradation experiments were also based on previous reports[30,32].The films cut in 6 mm diameter were used for enzymatic degradation experiments.After PET degradation experiments, the films were consecutively washed with 1% SDS, distilled water and pure ethanol.The dried films were subsequently observed by scanning electron microscopy (SEM) analysis.PET samples were coated with gold and SEM imaging was recorded on a field-emission scanning electron microscope (Apreo S LoVac, FEI, USA) operating at a beam-accelerating voltage of 5.00 kV under the high vacuum.
2.6.FITC dyeing experiment
To investigate whether the fusion of αS protein enhanced the binding affinity of the fusion enzymes to PET film, the proteins were labelled with FITC.In the modification,400 μl of FITC solution(1.6 mmol·L-1) and 1600 μl of enzyme solution (20 μmol·L-1) in 50 mmol·L-1sodium carbonate buffer at pH 9.0(20 μmol·L-1)were mixed and incubated at 4°C for 24 h[36].Unbound FITC molecules were removed by ultrafiltration and the labelled proteins were used for fluorescence microscopy.For the fluorescence observation, 200 nmol·L-1of FITC-modified PETase or its fusion enzyme was co-incubated with PET films for 1 h at 40 °C under shaking at 120 r·min-1.After the incubation,the PET films were rinsed with reaction buffer B (50 mmol·L-1glycine-NaOH, pH 9.0) to remove the unbound protein on the PET films.Then, PET film surface was observed with a fluorescence microscope(TE2000-U,Nikon,Tokyo,Japan).
3.Results and Discussion
3.1.Purification and activity assay of PETase and its fusion enzymes
αS protein domains from Homo sapiens were linked to the C terminal region of PETase, and six different linkers were utilized for the protein fusion (Fig.1).The specific linker sequences of the fusion proteins are provided in Table S1.It is known that GS(GGGGS) is a flexible linker while EK (EKKKA) is rigid [37].PETase and its fusion enzymes purified by IMAC were analyzed by SDSPAGE analysis.It is seen that the purified enzymes, marked by dashed rectangles in Fig.S2, were consistent with their corresponding theoretical molecular weights (MW) listed in Table S2.The MW at approximately 30 kDa and 15 kDa observed for the PETase and αS,respectively,were also in agreement with previous results[11,38].The protein yields of PETase and its fusion enzymes are presented in Fig.S3.It is interesting to see that the six fusion enzymes exhibited 1.3 to 2.9 times higher yield than PETase.The purified enzymes were utilized in subsequent experiments to explore their enzymatic properties.
As shown in Fig.2(a),PETase and all the fusion enzymes had the highest catalytic activity for BHET at 35 °C, indicating that the fusion of αS did not alter the optimum temperature of the hydrolase.Furthermore, the enzymatic activities of the six fusion enzymes were found to be 1.6 to 2.6 times higher than PETase at the optimum temperature of 35 °C (Fig.2(b)).Of the fusion enzymes, PETase-(GS)20-αS and PETase-(EK)20-αS with flexible and rigid linkers,respectively,presented similar enzyme activities,while the fusion enzyme with a mixed peptide linker of the same length, PETase-[(GS)(EK)]10-αS, displayed higher activity than its two counterparts.When doubling the mixed linker length, however, the fusion enzyme (PETase-[(GS)(EK)]20-αS) activity did not appreciably increase.

Fig.2.(a)Hydrolytic activities of PETase and its fusion enzymes at different temperatures using BHET as the substrate and(b)their activities at 35 °C.Reaction conditions:50 nmol·L-1 enzyme, 200 mg·L-1 BHET.Error bars represent SD values from triplicate experiments.
The results indicated that fusing the chaperone protein with all the tested linkers, either flexible or rigid, enhanced the hydrolytic activity of PETase towards BHET.One possible explanation for this enhancement is that the fusion of αS altered the structure of PETase,thereby allowing for better binding of the small molecular substrate to the active site,and consequently improving the enzymatic activities.This hypothesis will be further investigated through structural analysis.
3.2.Thermal stability of PETase and its fusion enzymes
The thermal stability of PETase and its fusion enzymes were evaluated by monitoring the residual activities following 6 h incubation at 40°C.Fig.3(a)shows that in the initial 30 min,the enzymatic activity of all the six fusion enzymes exhibited slower declines than PETase.Interestingly, after 6 h of incubation,PETase exhibited a residual activity of only 30.5%, whereas all the six fusion enzymes retained more than 60% of their initial activity,almost two times higher than the native enzyme.Notably, the activities of PETase-[(GS)(EK)]10-αS and PETase-[(GS)(EK)]20-αS exhibited <10% declines during the incubation, and kept 221%and 248% of the initial PETase activity (Fig.S4), respectively.

Fig.3.(a)Thermal stability of PETase and its fusion enzymes at 40°C.The residual activities were measured at different time periods and displayed as a relative percentage of their corresponding initial activities.(b) The Tm values of PETase and the fusion enzymes.
Fig.3(b)shows that the Tmvalues of the enzymes.It is seen that the Tmvalues of the fusion enzymes were 1.9 to 6.6°C higher than PETase.PETase-[(GS)(EK)]10-αS and PETase-[(GS)(EK)]20-αS showed the highest Tmvalues, consistent with the above kinetic thermal stability investigation.Notably,the stability and Tmvalues of PETase-(EK)20-αS with a rigid linker peptide were found to be the least improved in the six fusion enzymes.This could be attributed to that the chaperone protein fused through the rigid linker,(EK)20, might not effectively interacted with PETase to provide a positive effect on the overall structure of PETase as did through an equivalent length of a flexible linker,(GS)20.This conjecture will be further discussed by the structural analysis below.
Fluorescence spectroscopy was used to investigate the structural mechanism behind the stability enhancement of the fusion proteins.Intrinsic fluorescence emission was used to monitor alterations in the tertiary structure of PETase subsequent to its fusion with αS.As illustrated in Fig.S5, the fusion enzymes exhibited a blue-shift in the emission maxima (λmax) compared to PETase.This indicates that the protein molecules of the fusion enzymes have a more compact conformation, which could be responsible for their increased stability.Notably, PETase-(GS)6-αS, PETase-[(GS)(EK)]10-αS, and PETase-[(GS)(EK)]20-αS exhibited a greater degree of blue shift than the other enzymes (Table S3).These results are consistent with the stability measurements(Fig.3), in which the three enzymes showed higher stabilities or Tmvalues than the other counterparts.The observed enhancement in stability of the fusion enzymes could be attributed to the molecular chaperone effect of αS that stabilized its fusion partner,as it has been shown to inhibit the loss of ADH activity when co-incubated with ADH at 60°C[28].In addition,PETase-(GS)20-αS and PETase-(EK)20-αS showed the same degree of blue shift in the intrinsic fluorescence spectra, indicating that the fusion enzymes constructed with (GS)20and (EK)20linkers did not differ significantly in their effect/impact on the overall structure of PETase.The results imply that the above conjecture about the structure was disproved.Nonetheless, as seen in Fig.3, the stability and Tmvalue of PETase-(EK)20-αS were found to be inferior to that of PETase-(GS)20-αS.Therefore, a reasonable explanation for this is that the rigid long linker in PETase-(EK)20-αS significantly compromised the chaperone activity of αS, thereby weakening its protective effect on the protein.
There are eight tyrosine (Tyr, Y) and five tryptophan (Trp, W)residues (Fig.S6(a)) in the PETase structure (PDB ID 6EQE) [16].Specifically, Y87, W159, and W185 are located in the substrate binding pocket,where they are nestled deep within the hydrophobic core of the protein,and are known to play a pivotal role in facilitating substrate binding[12,32,39].The synchronous fluorescence spectra of Tyr and Trp exhibited a slight red-shift, as depicted in Fig.S7.The red-shift could be attributed to the relocation of Tyr and Trp residues from a buried hydrophobic environment to a solvent-exposed position [40].As a result of the conformational changes, the substrate-binding pocket of PETase (Fig.S6(a)) might shift from a relatively closed structure to a more open structure[41].This change might enhance the accessibility of the substrates to the active site, thereby improving the affinity of PETase for the hydrophobic substrates, leading to the increase of enzymatic activity.
It is noteworthy that because there are no Trp residues in αS(Fig.S6(b)), its fusion did not interfere with the Trp synchronous fluorescence spectra of the fusion enzymes.Moreover,the intrinsic fluorescence spectrum and the Tyr synchronous fluorescence spectrum of αS were exceptionally feeble (Figs.S5 and S7(a)), so their influences on the spectra of the fusion proteins could be ignored.The above results of intrinsic and synchronous fluorescence experiments(Figs.S5 and S7)are consistent with the findings of stability(Fig.3) and activity assays (Fig.2), providing a structural explanation for the increased stability and activity of the fusion enzymes.
3.3.Kinetics of enzymatic BHET hydrolysis
The enzymatic kinetics were studied by measuring the initial reaction rates at different BHET concentrations at 35 °C.The solubility limit of the BHET substrate in the reaction system restricted the enzymatic kinetic curves to a substrate concentration lower than 18.0 mmol·L-1.As demonstrated in Fig.S8, we were able to obtain linearly fitted kinetics curves for all the enzymes within the soluble substrate concentration range.Consequently, we were unable to obtain independent Michaelis-Menten equation parameters.At low substrate concentrations,when Kmgreatly exceeds the[S], the catalytic efficiency (kcat/Km) is an apparent second-order rate constant [42] and can be calculated directly from Eq.(4) [30],
where[E0]represents the enzyme concentration in the reaction system (50 nmol·L-1) and Vmax/Kmis the slope of the linearly fitted kinetic curve(Fig.S8).The calculated values of kcat/Kmare presented in Table 1.Notably,all the six fusion enzymes exhibited higher kcat/Kmvalues than PETase.In particular, the catalytic efficiency of PETase-[(GS)(EK)]10-αS and PETase-[(GS)(EK)]20-αS were 2.2 and2.4 times higher than PETase,respectively.These results are consistent with the activity assay shown in Fig.2.
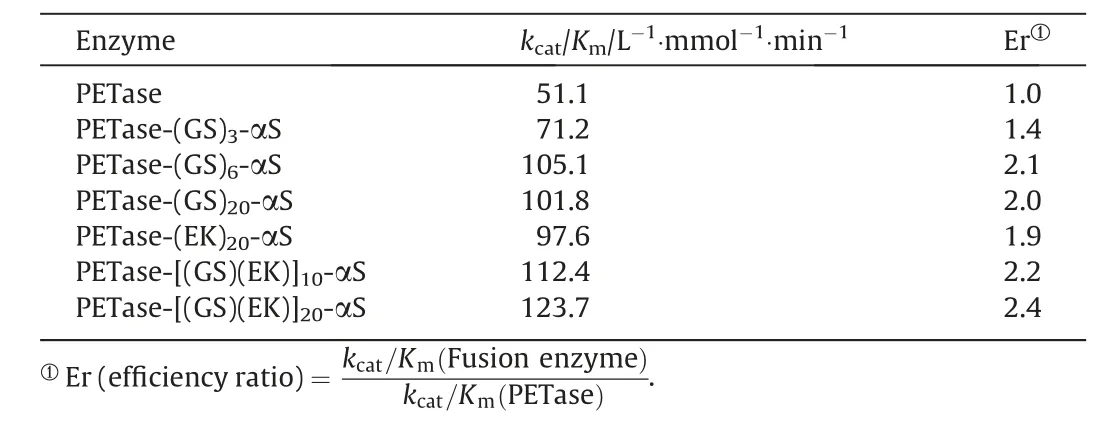
Table 1 Catalytic efficiency of PETase and its fusion enzymes
3.4.Degradation of PET films
To evaluate the practical application potential of PETase following fusion with αS, the resulting fusion enzymes were utilized to degrade Nongfu Mountain Spring bottle films (NfMSB-PET, crystallinity 32.3%, Fig.S9).The results, as depicted in Fig.4, demonstrated that PETase experienced a loss of activity within 3 d at 30°C.By contrast,all the six fusion enzymes exhibited an increasing trend in product release over a period of 5-6 d, and little increase was observed after the 6th day.Furthermore, all the six fusion enzymes exhibited higher product releases than PETase by the 7-d degradation.In particular, the product release of PETase-[(GS)(EK)]10-αS increased to 141.5 μmol·L-1after degrading NfMSB-PET at 30 °C for 7 d, which was 3.2 times higher than that of PETase.By contrast, however, the two fusion enzymes with the same linker length but flexible (PETase-(GS)20-αS) and rigid(PETase-(EK)20-αS) liners showed much lower degradation, and the degradation efficiency of PETase-(EK)20-αS was lower than that of PETase-(GS)20-αS,probably because the rigid long linker peptide limited the active site of PETase in contacting well with the substrate PET.Moreover, doubling the mixed linker length (i.e.,PETase-[(GS)(EK)]20-αS) significantly compromised the degradation efficiency.

Fig.4.The efficiency of degradation of Nongfu Mountain Spring bottle films by PETase and its fusion enzymes at 30°C.The released compounds were the sum of MHET and TPA at different treatment time.
Besides,we found that PETase had significantly higher degradation efficiency than its fusion enzymes at 30°C in the first 3 d.The reason for this phenomenon was not clear.We speculate that the adsorption binding mediated by αS in the fusion enzyme (as shown in Fig.S12) might have inhibited the contact between the enzyme active site and the PET film in the initial stage, thereby limiting the degradation capacity of the fusion enzyme in the early stage.However,the orientation of the bound fusion enzyme on the surface via αS would continuously change to make a reasonable contact between the active site and the PET surface.As a result,the degradation capacity of the fusion enzyme could be fully exploited after the favorable orientation was taken, and its degradation efficiency increased significantly over time.In contrast,free PETase did not involve this process and could degrade PET by free contact with the surface.Therefore,PETase exhibited a higher product release than the fusion enzymes in the first few days.At higher temperature (40 °C, Fig.S10), the delay in the increase of the degradation capacity of the fusion enzymes were not observed.This implies that the orientation adjustment of the bound fusion enzymes on the surface to take a favorable orientation needed much shorter time at this high temperature than that at 30 °C.
The surface morphology of the degraded films was analyzed using SEM, as illustrated in Fig.5.It was observed that the NfMSB-PET surfaces hydrolyzed by the fusion enzymes displayed deeper and more roughness than that by PETase, and gradually developed porous structures.Remarkably, PETase-[(GS)(EK)]10-αS showed the largest degradation of the NfMSB-PET films among the enzymes,leading to the appearance of large and deep irregular pores on the surface of the films.This is consistent with the product releases shown in Fig.4.However, as shown in Figs.S10 and S11, the fusion enzymes with a short flexible linker (PETase-(GS)3-αS) or a rigid linker (PETase-(EK)20-αS) did not show any enhanced performance over native PETase at 40 °C.Although the other four fusion enzymes were more effective than native PET,their overall degradation at this temperature was not as effective as at 30 °C.Despite this, the product release from PETase-[(GS)(EK)]10-αS degradation was also the highest, reaching 5.1 times higher than that of native PETase in 4 d (Fig.S10).

Fig.5.SEM images of the enzyme-treated PET film surfaces at 30°C.The Nongfu Mountain Spring bottle films(diameter,6 mm)were incubated with 200 nmol·L-1 PETase or its fusion enzymes in reaction buffer B (50 mmol·L-1 glycine-NaOH, pH 9.0) for 7 d at 30 °C.Corresponding films treated with buffer B were set as controls.
Overall, of the six fusion enzymes, PETase-[(GS)(EK)]10-αS demonstrated the highest efficiency in degrading PET (Fig.4 and Fig.S10).To evaluate the enzyme efficiency based on both enzyme production and activity, the product accumulations were calculated based on enzymes produced with 1-L bacterial culture(Fig.S3) and the enzyme efficiency in product releases in the degradation of PET films(Fig.4 and Fig.S10).The results of the calculation for the accumulation of degradation products after 7 d are listed in Table S4.It is revealed that PETase-[(GS)(EK)]10-αS exhibited the highest improvement in product accumulation, 7.0 times higher at 30 °C and 10.7 times higher at 40 °C than PETase.Based on the aforementioned activity and stability experiments, PETase-[(GS)(EK)]10-αS exhibited superior performance to the other fusion enzymes, and had a high enzyme yield (Fig.S3), which were the causes for its superiority over other counterparts.
In summary,all the six fusion enzymes demonstrated enhanced performance in terms of enzymatic activity and stability, as compared to PETase.(Figs.2 and 3).Hence,αS can be considered as a platform chaperone for building fusion enzymes of improved activity and stability.Moreover, it has been proven that αS domain helped adhesion of the fusion enzymes to PET films during PET degradation, as evidenced by the increased number of PETase molecules per PET surface unit (Fig.S12).This observation was consistent with literature which reported that the C-terminal domain of αS could bind to PET surface[28].This binding increased the affinity of the fusion enzymes for the substrate,which,together with the increased activity and stability of the fusion enzymes,led to the increases in PET degradation efficiency(Figs.4 and 5).Compared with the limited enhancement effect of the fusion of αSP reported by earlier [29], αS fusion can bring a greater degree of degradation efficiency improvement due to the collective effects of activity enhancement (Fig.2), stability improvement (Fig.3)and increase in affinity for PET films (Fig.S12).The linker peptide also played a critical role in the enzymatic hydrolysis.A linker of suitable flexibility and length would be crucial to keep the accessibility of the active site of the PETase domain in a fusion enzyme.As two frequently used linkers, GS is flexible while EK is rigid [37].A flexible linker of(GS)6was suitable to keep a high efficiency of the fusion enzyme of PETase-(GS)6-αS, while increase in the linker length to 100 amino acid residues ((GS)20) did not further significantly increase the catalytic efficiency of the fusion enzyme of PETase-(GS)20-αS (Fig.4 and Fig.S10).For the mixed linker with both GS and EK, however, the fusion enzyme with the linker of 100 amino acid residues ([(GS)(EK)]10) possessed higher catalytic efficiency than the fusion enzyme of the same flexible linker length, PETase-(GS)20-αS, both for BHET (Fig.2) and PET (Fig.4 and Fig.S10).
Indeed,linker peptides have been demonstrated to significantly impact on the catalytic performance of fusion proteins[24,43-45].When constructing fusion proteins, there are numerous potential linker sequences available, and the optimal choice for a given fusion protein can depend on various factors, including the size,conformation, and function of the two proteins to be fused.Currently, the selection of appropriate linkers for the construction of fusion proteins continues to be largely reliant on a trial-and-error strategy [21].This typically entails testing linker peptides of varying lengths, compositions, and sequences to identify the optimal combination.Furthermore,the stability and solubility of the resulting fusion protein can be affected by the linker peptide.Thus, it is essential to optimize the linker peptide to achieve the best performance of the fusion protein.Our results have indicated that PETase-[(GS)(EK)]10-αS, a fusion protein with equal ratio of mixed GS and EK linker exhibited the highest performance among the fusion enzymes.This proved that the linker ([(GS)(EK)]10) of suitable flexibility and length maximized its catalytic efficiency for PET degradation.Because of the diversity of linker peptides [37],optimization of the linker peptide by more extensive experiments has the potential to further enhance the catalytic performance of the fusion enzyme.
4.Conclusions
In light of the adhesive characteristics and chaperone activity of αS protein,a fusion approach was proposed to attach αS protein to the C-terminal of PETase through linker peptides of varying lengths and flexibilities.Results showed that all the six fusion enzymes exhibited improved catalytic activity and thermal stability, which was attributed to the αS fusion-induced changes in the tertiary structure of the enzyme.The results suggested that αS can be a chaperone domain for building fusion enzymes of improved activity and stability.Furthermore, FITC staining experiments confirmed the binding effect on PET films by the αS fusion.It was shown that the linker peptides of different lengths and compositions brought about different catalytic performance enhancements.Among the PETase fusions, PETase-[(GS)(EK)]10-αS demonstrated the most efficient catalytic performance for BHET and PET.Thus,this work has demonstrated the efficiency of the chaperone protein αS for building fusion enzymes of improved activity and stability,and it is more suitable for enzymes (e.g., PETase) that need to increase its affinity for substrate by such a fusion.Linker optimization would further increase the chaperone power of αS.Recently,several PETase variants with much higher catalytic performance than the wild-type PETase have been developed [46-48].It would be interesting to extend the fusion strategy to the PETase variants to investigate whether αS could increase their catalytic activities,particularly PET degradation performances.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2018YFA0900702).
Supplementary Material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cjche.2023.06.015.
 Chinese Journal of Chemical Engineering2023年12期
Chinese Journal of Chemical Engineering2023年12期
- Chinese Journal of Chemical Engineering的其它文章
- Intrinsic kinetics of catalytic hydrogenation of 2-nitro-4-acetylamino anisole to 2-amino-4-acetylamino anisole over Raney nickel catalyst
- Experiments and model development of p-nitrochlorobenzene and naphthalene purification in a continuous tower melting crystallizer
- Influence of water vapor on the separation of volatile organic compound/nitrogen mixture by polydimethylsiloxane membrane
- Mass transfer mechanism and relationship of gas-liquid annular flow in a microfluidic cross-junction device
- Enhanced photocatalytic activity of methylene blue using heterojunction Ag@TiO2 nanocomposite: Mechanistic and optimization study
- Comparative analysis on gas-solid drag models in MFIX-DEM simulations of bubbling fluidized bed