
特发性肺间质纤维化合并原发性肺滑膜肉瘤1例并文献复习
2023-02-14 08:22陶会会谭恩丽李鑫刘晓菊董静包海荣
临床肺科杂志 2023年2期
陶会会 谭恩丽 李鑫 刘晓菊 董静 包海荣
本文报道1例特发性肺间质纤维化(idiopathic pulmonary fibrosis, IPF)合并原发性肺滑膜肉瘤(primary pulmonary synovial sarcoma, PPSS),就其临床特征、影像、诊断与鉴别诊断分析如下。
病例资料
患者,男,66岁,因“间断咳嗽、气短4年,加重伴右侧胸痛10天”入院。患者于4年前无明显诱因出现咳嗽、气短,胸部CT(2017年11月22日)示双肺间质纤维化(图1a),肺功能示轻度限制性通气功能障碍,弥散功能中度下降(DLCO% 53.3%)。诊断为IPF,间断口服N-乙酰半胱氨酸(N-acetyl cysteine, NAC)治疗。于1年前咳嗽、气短逐渐加重,复查胸部CT(2020年10月24日)示双肺间质纤维化加重(图1b)。复查肺功能示通气功能指标变化不大,DLCO% 降至38.1%,因经济原因未正规抗纤维化治疗,继续口服NAC。于入院前10天因咳嗽、呼吸困难加重,伴右胸撕裂样疼痛住院。患者既往有糖尿病和冠状动脉粥样硬化性心脏病病史,吸烟史40年。入院查体:生命体征平稳,体型消瘦,口唇发绀,右下肺叩诊浊音,听诊右下肺呼吸音低,双下肺可闻及Velcro啰音,可见杵状指。血气分析(吸氧5L/min)示PH 7.46,PO252 mmHg,PCO236.5 mmHg,SO288.5%。复查胸部CT(2021年10月13日)示肺间质纤维化进一步加重,新增右肺下叶混杂密度团片影,右侧胸腔积液(图1c、d)。CT胸痛三联血管成像示右冠脉中段混合斑块,管腔重度狭窄,主动脉弓及分支粥样硬化,肺动脉高压,右心增大,未见肺栓塞及主动脉夹层。行右侧胸腔穿刺术,抽出暗红色血性胸水300mL,化验胸水常规和生化符合渗出液改变,胸水癌胚抗原17.9ng/mL,化验血常规、肝、肾功能均正常,自身抗体全项均阴性。CT引导下肺穿刺活检病理:未见正常肺组织,瘤细胞梭形,细胞核短梭形、卵圆形,可见病理性核分裂象,呈束状、弥漫排列,其内可见少量巨核、多核、奇异核瘤细胞,局灶可见少量上皮样细胞,呈巢状、条索样排列。免疫组化显示vimentin(3+),TLE(+),EMA(+),bcl-2(2+),CD99(+),TTF-1(-),CKP(+),CR(-),Desmin(-),S-100(-),Ki67(40%)(图2)。诊断为肺滑膜肉瘤。头颅MRI平扫、腹部增强CT扫描、全身ECT骨显像均未见明显异常。明确诊断为IPF合并PPSS。经过多学科诊疗(Multi-disciplinary Treatment, MDT)讨论,患者一般状况差,体力状况(performance status, PS)评分2分,不能耐受抗肿瘤化疗及放疗,给予口服安罗替尼抗肿瘤,同时辅以氧疗、引流胸腔积液及针对糖尿病和冠状动脉粥样硬化性心脏病基础病治疗1周后好转出院。出院后患者仍气短、胸痛,遂再次住院,复查胸部CT(2021年11月16日)示右肺下叶混杂密度团块影较前明显增大(图1e、f)。再次MDT讨论后,建议患者继续口服安罗替尼治疗。但患者病情持续进展,1月后死亡。

图1 胸部HRCT
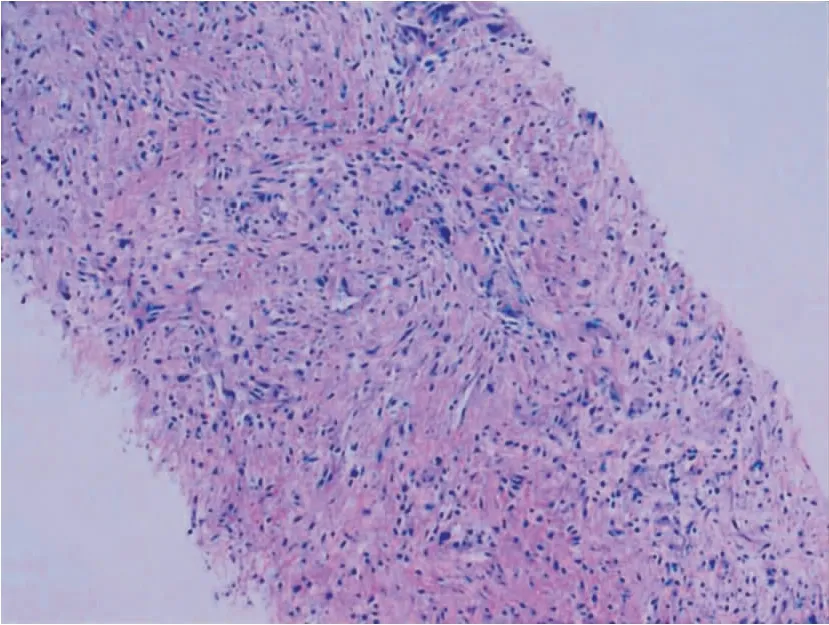
图2 经皮肺穿刺活检组织病理(HE×100)
讨 论
IPF是一种病因不明的慢性、进行性、纤维化性间质性肺炎,好发于老年人,起病隐匿,主要表现为干咳和进行性加重的呼吸困难。胸部高分辨率CT(high resolution CT, HRCT)和组织病理学特征性表现为普通型间质性肺炎(usual interstitial pneumonia, UIP),是诊断IPF的金标准[1]。IPF需要与其他可以引起UIP型纤维化的间质性肺疾病鉴别。本例患者老年男性,有长期大量吸烟史,无职业及环境因素暴露史,无长期服药史,以咳嗽和呼吸困难为主要症状,无结缔组织疾病相关症状,查体双下肺可闻及Velcro啰音,可见杵状指,自身抗体检查均阴性,肺功能示限制性通气功能障碍,弥散功能下降,胸部HRCT示双肺胸膜下及下叶为著的网格及蜂窝样改变,符合IPF的诊断。
IPF预后差,确诊后中位生存期仅2~3年[2],5年生存率不足50%[3]。IPF目前尚不能治愈,治疗目的是延缓疾病进展,改善患者的生活质量,延长生存时间。主要治疗方法包括非药物治疗(如氧疗、肺康复训练、肺移植等)和抗纤维化药物治疗。指南推荐吡非尼酮和尼达尼布抗纤维化治疗,有条件不推荐NAC治疗IPF[4]。本例患者因经济条件所限,未能使用抗纤维化药物,而单纯口服NAC治疗,肺间质纤维化持续进展。
PPSS是一种罕见的,高度恶性的软组织恶性肿瘤,临床表现无特异性,主要为咳嗽、咳痰、胸痛、咯血、呼吸困难等,部分患者可发生顽固性自发性气胸和肺大疱,也有患者无任何症状[5]。PPSS胸部CT多表现为边缘清晰的单发肿块,直径一般>5cm,呈类圆形或不规则形,伴坏死或囊性强化,侵犯胸膜或胸壁时可引起胸腔积液[6]。SS的组织学分类主要有低分化型、双相型(由上皮细胞和梭形细胞构成)、单相纤维型(由梭形细胞构成)和单相上皮细胞型(由上皮细胞构成)4类,以双相型和单相纤维型最常见。PPSS通常表达EMA、Bcl-2、CD99、CK和Vimentin,不表达SMA、Desmin和S-100[7]。PPSS需要与肺转移性滑膜肉瘤、平滑肌肉瘤、纤维肉瘤、恶性周围神经鞘瘤等疾病相鉴别。肺转移性滑膜肉瘤存在原发病灶,通常可以触及肿块,伴触痛,CT多表现为大小不等、边缘清晰的圆形或类圆形结节影。平滑肌肉瘤的梭形细胞胞浆伊红染色深,免疫组织化学染色 SMA和DeSmiri阳性。纤维肉瘤的梭形细胞呈束状交织排列,上皮标记阴性。恶性周围神经鞘瘤的梭形细胞呈波浪状,免疫组化S-100阳性,上皮标记阴性[8]。本例患者主要症状为咳嗽、气短、胸痛,容易被 IPF的症状掩盖。胸部CT右肺下叶可见巨大混杂密度团块影,右侧包裹性胸腔积液,肺组织病理学及免疫组化结果符合SS,故可明确诊断为PPSS。
PPSS的治疗方案主要根据肿瘤分期选择。对于无淋巴结转移、肿瘤<5cm的患者以手术切除为主;对于不能手术的晚期患者选择化疗、放疗、分子靶向(如安罗替尼)及免疫治疗(NY-ESO-1)[9]。PPSS化疗方案主要是多柔比星联合异环磷酰胺,其他化疗药物还有蒽环类药物如阿霉素等。化疗可以使肿瘤缩小并治疗潜在的微小转移灶,提高肺SS患者的生存率[10]。安罗替尼是一种多靶点的受体酪氨酸激酶抑制剂,通过抑制成纤维细胞生长因子受体(fibroblast growth factor receptor, FGFR)、血管内皮生长因子受体(vascular endothelial growth factor receptor, VEGFR)及血小板衍化生长因子受体(platelet-derived growth factor receptor, PDGFR),抑制肿瘤血管生成及生长,通过抑制干细胞因子的受体C-Kit,干扰肿瘤细胞的正常生长[11]。本病例患者因不能耐受化疗和放疗,采用安罗替尼抗肿瘤治疗,但疗效差,患者很快死亡。
研究表明IPF患者发生肺恶性肿瘤的风险比正常人高7%~20%,多见于有吸烟史并伴有肺气肿的患者[12]。IPF与肺恶性肿瘤有多种共同的分子和细胞过程,如成纤维细胞激活和不受控制的增殖、成纤维细胞间充质转化、内质网应激、生长因子表达的改变、氧化应激以及遗传和表观遗传变异等[13]。与单纯IPF患者相比,IPF合并肺恶性肿瘤患者的平均生存时间更短[14]。Kato等[15]报道IPF患者诊断肺恶性肿瘤后1年、3年和5年的全因死亡率分别为53.5%、78.6%和92.9%。IPF患者最常见的肺恶性肿瘤是鳞状细胞癌,其次是腺癌,也有报道大细胞肺癌和小细胞肺癌[16-17]。尚未见IPF合并PPSS的报道。本例患者4年前诊断IPF,但未及时抗纤维化药物治疗,未定期评估,导致IPF逐渐进展,且未及时发现合并肺恶性肿瘤,失去了最佳治疗时机。
综上所述,IPF合并PPSS患者通常起病隐匿,肿瘤症状容易被 IPF的症状掩盖,治疗手段有限,而且放、化疗和手术治疗都可能导致IPF进展,所以IPF合并PPSS时治疗更加困难。应对IPF患者定期评估,早诊断、早治疗,及早发现合并症或并发症,以延缓疾病进展,提高患者的生存时间。
猜你喜欢
舰船科学技术(2022年10期)2022-06-17
中国药学药品知识仓库(2022年8期)2022-05-09
建材发展导向(2021年14期)2021-08-23
电脑报(2021年49期)2021-01-06
中国临床医学影像杂志(2019年4期)2019-06-18
癌症进展(2018年14期)2018-12-31
西安建筑科技大学学报(自然科学版)(2016年5期)2016-11-10
中国组织化学与细胞化学杂志(2016年3期)2016-02-27
磁共振成像(2015年1期)2015-12-23
郑州大学学报(医学版)(2015年1期)2015-02-27
