
The structure-sensitive of Cu catalyst for furfural conversion to furfuryl alcohol:A theoretical study
2023-02-07 08:01WANGGuiruZHICuimeiYANGWen
燃料化学学报 2023年2期
WANG Gui-ru,ZHI Cui-mei,YANG Wen
(1.College of Chemical and Biological Engineering, Shanxi Key Laboratory of High Value Utilization of Coal Gangue,Taiyuan University of Science and Technology, Taiyuan 030024, China;2.Shanxi Key Laboratory of Metal Forming Theory and Technology, School of Material Science and Engineering, Taiyuan University of Science and Technology, Taiyuan 030024, China)
Abstract: The structure-sensitive of Cu catalyst for furfural hydrogenation to furfuryl alcohol was explored by employing Cu(111) and Cu(211) model systems.Herein,the adsorption behavior of reactants and intermediates,and the possible reaction mechanism of furfuryl alcohol formation were investigated.For furfuryl alcohol formation,the preferred pathway is F-CHO +2H→F-CH2O+H→F-CH2OH,in which the second H addition is the rate-determining step.Meanwhile,Cu(211) surface exhibits higher activity to furfuryl alcohol formation than that on Cu(111) surface.According to our analysis,the undercoordinated sites on the Cu(211) surface could facilitate H2 dissociation and stabilize the adsorbed furfural,thereby promoting the furfural hydrogenation and the furfuryl alcohol formation.This work provides a feasible approach for regulating the catalytic activity and selectivity in furfural conversion.
Key words: Cu catalysts;structure-sensitive;furfural hydrogenation;furfuryl alcohol
The global climate changes and the diminishing reservation of fossil resources have received considerable attention.Biomass are abundant and effective carbon-sustainable sources,and they are also regarded as important alternative energies.Furfural,as one of the promising functionalized biomass-derived molecules,can be obtained from hemicelluloses[1-8].Due to the high reactivity of furfural,it can be used to synthesize a series of chemical intermediates and apply in downstream production[9-10]. Catalytic selective hydrogenation of furfural into furfuryl alcohol has attracted great interest in terms of economics and safety.Conversion of furfural into furfuryl alcohol has generally been performed on metal catalysts,such as noble metal Pd[11],Pt[12],and Ru[13]-based system.Unfortunately,the high cost and poor selectivity of these catalysts preclude them from the industrial application.Cu-Cr catalysts are highly active and selective in industrial furfural hydrogenation,but they can cause environmental pollution because of the toxicity of Cr6+[14,15].To overcome these issues,a great effort has been taken to synthesize environmentally friendly Crfree catalysts for furfuryl alcohol production,involving Cu-Pd,Pt or Ni alloy.It has been found that the catalyst surface structures (terraces,steps,and corners)can impose a key effect on product distribution in Ni[16,17]and Pd[18]systems.For example,Wang et al.[18]believed that the activation of furan ring of furfural and the hydrogenation of C=O group are carried out at terrace and step edge of Pd,respectively.Recently,Zhang’group stated that high coordinated Ni surface is responsible for the formation of tetrahydrofurfuryl alcohol,whereas more 2-methylfuran are generated on low coordinated Ni surface in furfuryl alcohol conversion[17].Up to now,the mechanism for furfuryl alcohol formation from furfural conversion over modifying Cu catalysts is not fully understood.
This work employs Cu(111) and Cu(211) surfaces to address structure sensitive of Cu catalyst in furfural conversion to furfuryl alcohol.Cu(111) is the most stable surface[19-21],while Cu(211) surface consisting(100)-type step and (111) terrace exhibits higher reactivity[22-26].The adsorption behaviors of reactant of furfural (F-CHO),the main intermediate (F-CH2O,FCHOH) and the goal product of furfuryl alcohol (FCH2OH),and the possible reaction mechanism were systematically investigated by Density Functional Theory (DFT).This study opens up a strategy of modulating the structure of metal surface for the selective hydrogenation of furfural.
1 Calculation details
As shown in Figure 1,5 × 5 Cu(111) and 2 × 4 Cu(211) surfaces were constructed to represent the Cu catalysts with different structures.Cu(111) surface consists of four atom layers;the bottom two layers were frozen,whereas the remaining atoms and reacting molecules were allowed to relax.For the stepped Cu(211) surface,eight atom layers were modeled,and the top four layers and the adsorbates were allowed to relax,while the rest atoms were fixed.A vacuum of 15 Å was imposed to avoid the interaction between the adjacent slabs.2 × 2 × 1 Monkhorst-Pack mesh was used for the Brillouin-zone integrations in the calculation.

Figure 1 Possible adsorption sites on Cu(111) and Cu(211)surface from top and side view
All periodic DFT calculations were implemented in the Vienna Ab initio Simulation Package (VASP)software[27].The Perdew,Burke,and Ernzerhof (PBE)functional was utilized to compute the exchangecorrelation energy[28].To address the van der Waals(vdW) interactions between the adsorbate and the surface,the semiempirical Grimme’s D3 correction[29,30]was adopted.A plane-wave basis set with a cutoff of 400 eV in combination with the projected augmented wave (PAW) method was used[31,32].The geometry optimization was performed with force threshold of 0.05 eV/Å and the energy threshold of 10-5eV.The transition states were searched by the climbing image nudged elastic band (CI-NEB) method[33],and they were further optimized by the dimer method[34,35].Stable reactants,transition states and products configurations were verified by vibrational frequencies.
The adsorption energyEadsis calculated by eq.(1):
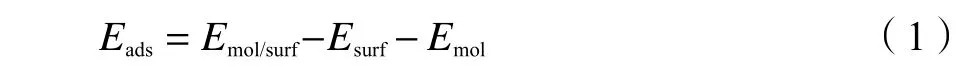
whereEmol/surfis the total electronic energy of adsorbate adsorbed on the surface,Emol/surfandEmolare the total electronic energies of the pure Cu surface and molecule in the gas phase,respectively.The more negative valueEadsis,the more the stable adsorbate structure is.
The activation barrier (Ea) is defined as the energy differences between the transition state (TS) and initial state (IS),whereas for the reaction energy (ΔEr),it is the difference between the final state (FS) and initial state (IS) energies,calculating by eq.(2) and (3):

Thed-band center (εd) is usually used to predict the interaction of adsorbate and metal surface,which is calculated by eq.(4):

whereEis the energy of Fermi level,and ρdrefers to the projected state density (PDOS) of the atomd-band of catalyst surface.
2 Results and discussion
2.1 Adsorption of intermediates
2.1.1 Cu(111) surface
The adsorption sites (e.g.top,bridge,fcc,and hcp site)[36]on Cu(111) surface and the stable adsorption configurations of intermediates as well as the key geometric parameters were shown Figures 1 and 2,and Table 1,respectively. Furfural is favorable for adsorption in a flat configuration with the O7atom of CHO group binding with the Cu atom,according to previous reports[36-38].The calculated adsorption energy is -0.70 eV. Besides,the adsorption of the intermediates involving in furfuryl alcohol formation was also taken into account.
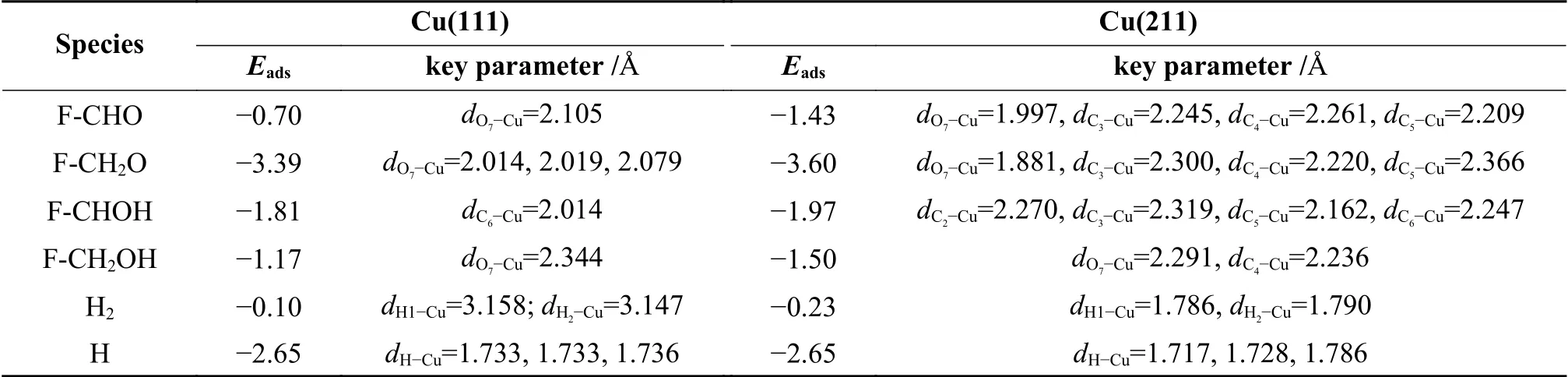
Table 1 Adsorption energies and key geometrical parameters of various intermediates on Cu(111) and Cu(211) surface
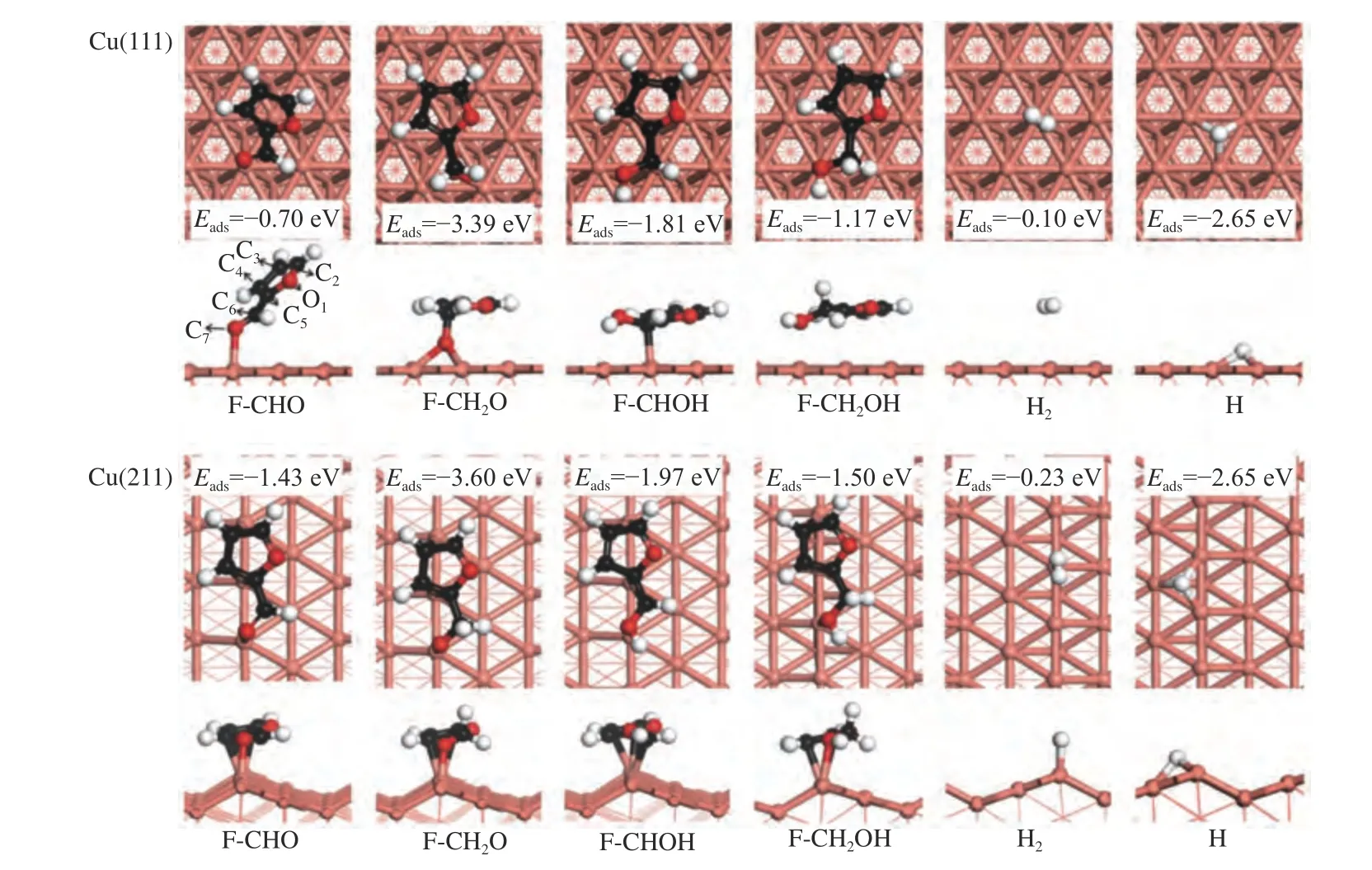
Figure 2 Intermediates involved in furfuryl alcohol formation via furfural hydrogenation on Cu(111) and Cu(211) surface
Furfuryl alcohol tends to adopt a flat configuration,similar as that of furfural.The O7-Cu distance is 2.344 Å and the corresponding adsorption energy for furfural alcohol is -1.17 eV. The intermediates of F-CH2O and F-CHOH favor adsorbing at the fcc and top site via O7atom and C6atoms,with the adsorption energies of -3.39 and -1.81 eV,respectively.H atom can be adsorbed either hcp(-2.65 eV) or fcc site (-2.66 eV),and the calculated results are in line with previous reported value (-2.54 and -2.55 eV)[36].
2.1.2 Cu(211) surface
In order to understand the influence of surface structure on the adsorption,the adsorption behavior of reactant and intermediates was further investigated on Cu(211) surface.On Cu(211) surface,top,bri,hcp and fcc at the step edge,and hollow site at the (100) step were considered.Furfural tends to use O7atom to adsorb at the step edge of the Cu top site,along with the binding of C3,C4and C5atoms of furanic ring with the Cu surface.The O7-Cu,C3-Cu,C4-Cu and C5-Cu bond lengths are 1.997,2.245,2.261 and 2.209 Å,respectively.The O7-Cu distance (dO7-Cu) is obviously decreased from 1.997 Å on Cu(211) surface to 2.105 Å on Cu(111) surface,thereby making the adsorption energy become higher on Cu(211) (-1.43 eV) than on Cu(111) surface (-0.70 eV).
For F-CH2O,it is adsorbed at the step Cu siteviaO7of CH2O group and the C3,C4and C5atoms of furanic ring,with the distance of O7-Cu,C3-Cu,C4-Cu and C5distances of 1.881,2.300,2.220 and 2.366 Å,respectively.The corresponding adsorption energy is -3.60 eV.The F-CHOH favors to adsorb at the Cu step edge via C6atom of CHO group,and the C2,C3and C5atoms of furanic ring.The C6-Cu bond length and the calculated adsorption energy are 2.247 Å and -1.97 eV,respectively.Furfuryl alcohol (FCH2OH) is adsorbed at the step edge of Cu atom with the adsorption energy of -1.50 eV,which is stronger than that on Cu(111) surface.The adsorption of H atom at hcp and fcc site shows similar energy to that adsorption on Cu(111) surface.
Based on the above results,it could be found that the intermediates in the formation of furfuryl alcohol are inclined to adsorbing at the step edge of Cu(211)surface.The different adsorption energy and adsorption configurations of these intermediates on Cu(211)surface and Cu(111) surface indicate that geometric and electronic properties of various Cu surfaces can affect the production distribution.Thus,the mechanism for furfural conversion to furfuryl alcohol on Cu(111)and Cu(211) surface was further explored in the following section.
2.2 H2 dissociation and diffusion
Since H2activation is indispensable in the process of furfural conversion to furfuryl alcohol,H2dissociation and diffusion were investigated (Figure 3).The adsorption of H2on Cu(111) is weak with the adsorption energy of only -0.10 eV,being in line with the previous reported results ((-0.08)-(-0.07) eV[36,39]).The step edge of Cu(211) surface effectively promotes the adsorption of H2with the adsorption energy increasing to -0.23 eV.Meanwhile,the H-H bond length of adsorbed H2on Cu(211) surface (0.803 Å) is larger than on Cu(111) (0.753 Å),although both of them are higher than that of the gas H2molecule (dH-H=0.750 Å[40]).The activation energy of H2dissociation on Cu(111) surface is 0.54 eV,which is consistent with the theoretical value reported in the previous work[36,39].However,it is decreased to 0.46 eV on Cu(211)surface,with the reaction energy of -0.50 eV.Thus,H2is preferentially dissociated into two H atoms on Cu(211) surface than on Cu(111) surface.After H2dissociation,the diffusion of the generated H atom on Cu(111) and Cu(211) surface was further illustrated.The migration of H atom from the hcp site to the fcc site of Cu(111) surface or of Cu(211) surface requires a low energy barrier of 0.14 and 0.17 eV,respectively,indicating that H atom diffusion is quite facile on both surfaces.As a result,the low dissociation and diffusion energy barrier could promote the subsequent hydrogenation and hydrogenolysis reaction[41].
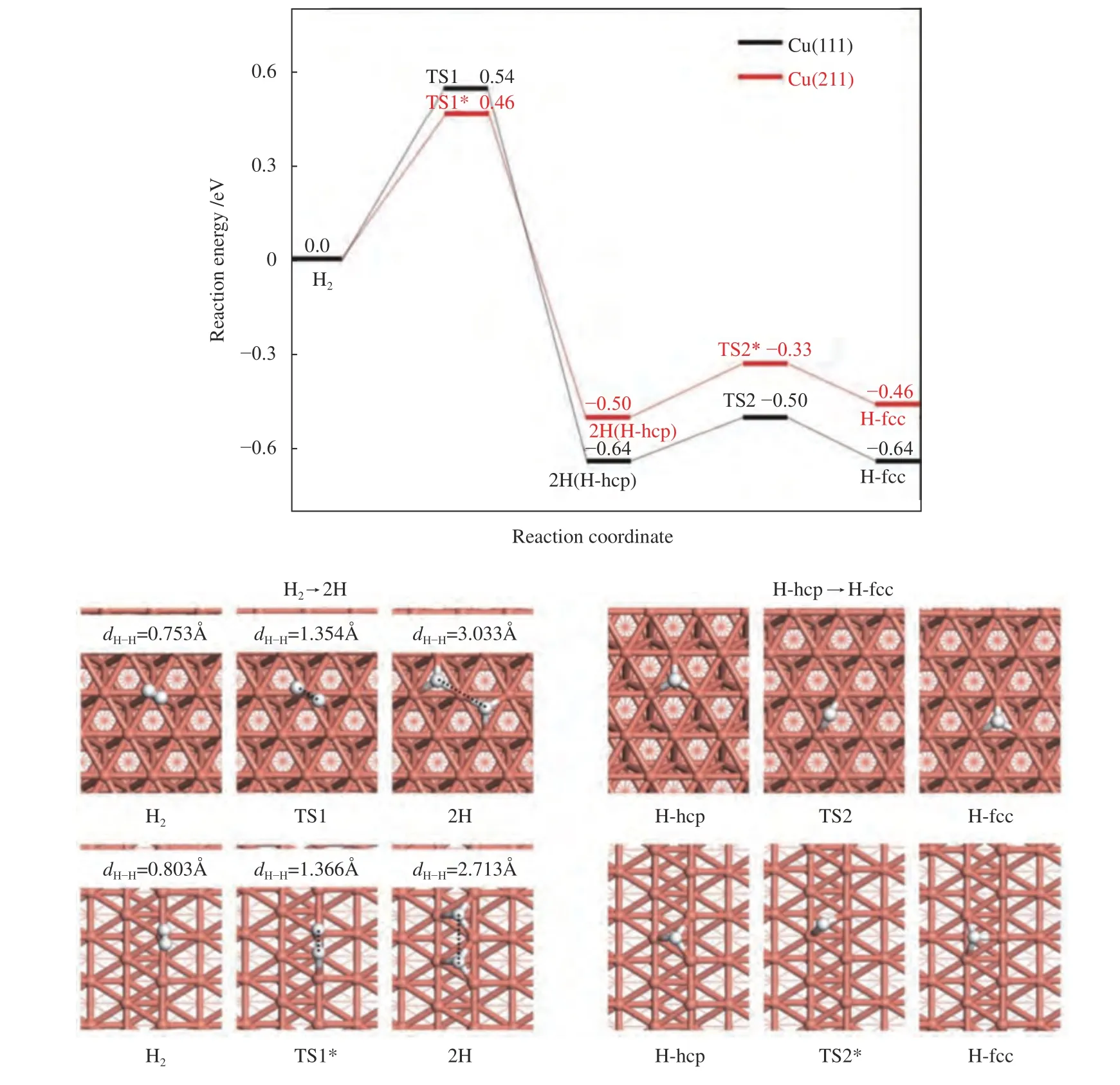
Figure 3 Reaction pathway for H2 dissociation and diffusion on Cu(111) surface and Cu(211) surface
2.3 Mechanism of furfural conversion to furfuryl alcohol
With the acquisition of the stable adsorption configuration,the possible reaction mechanism of furfuryl alcohol formation over Cu(111) surface and Cu(211) surface was then unraveled. The corresponding activation barriers and reaction energies were displayed in Table 2.
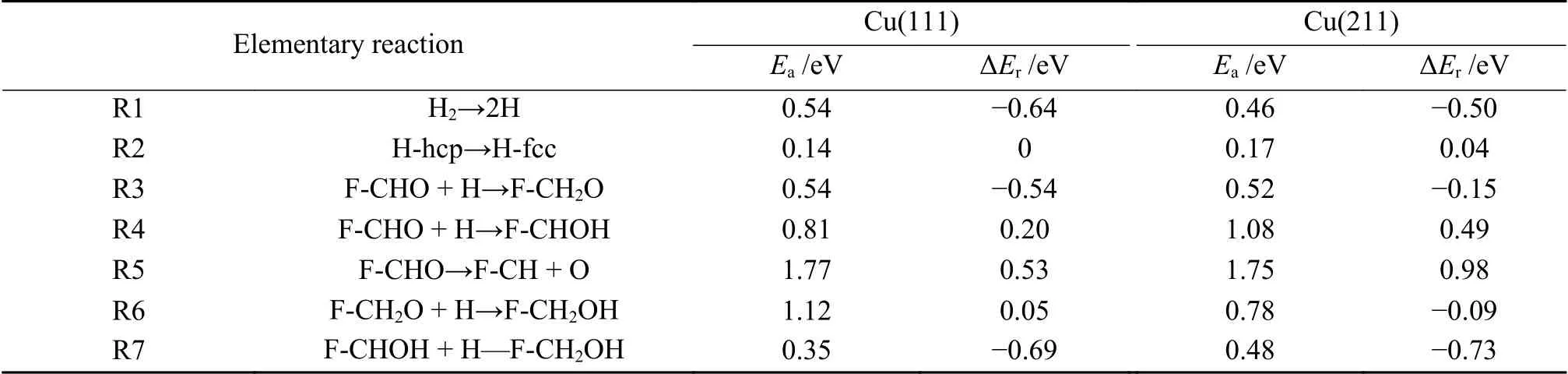
Table 2 Activation barriers and reaction energies of various elementary reactions involving in furfural alcohol formation on Cu(111) and Cu(211) surface
2.3.1 Mechanism on Cu(111) surface
Two possible pathways have been proposed for furfural hydrogenation to furfuryl alcohol,as shown in Figure 4.
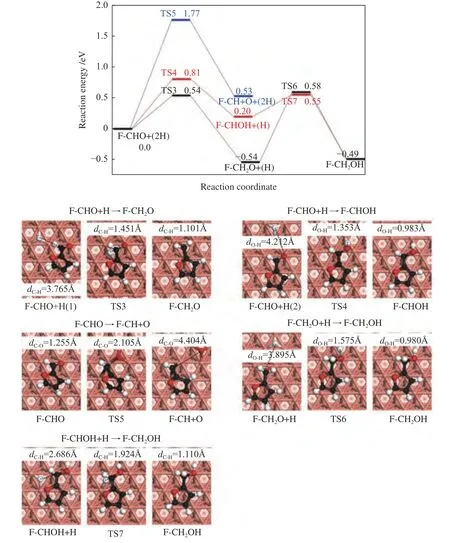
Figure 4 Reaction potential energy profile of furfuryl alcohol formation on Cu(111) surface
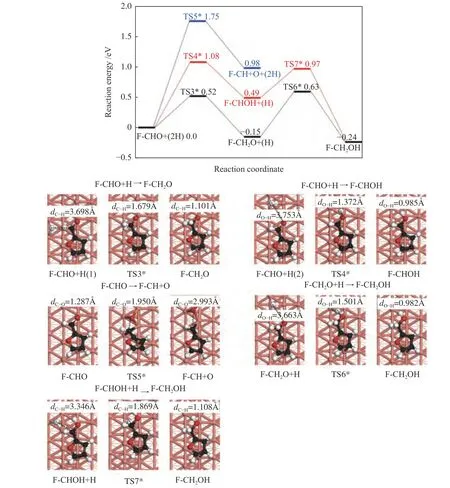
Figure 5 Energy profile of furfural conversion to furfuryl alcohol on Cu(211) surface
Furfural can be hydrogenated by C6or O7to generate alkoxide intermediate (F-CH2O) or hydroxyalkyl (F-CHOH). The first hydrogenation mode occurs at the C6atom of C=O group,which requires the energy barrier and the reaction energy of 0.54 and -0.54 eV,respectively.In TS3,C6-H distance is decreased to 1.451 Å.For the formation of F-CHOH,the hydrogen atom will firstly attack to O7atom,and this process is endothermic by 0.20 eV,with a barrier of 0.81 eV.In the TS4,H is located at the top site of Cu with the O7-H bond length of 1.353 Å.The calculated results suggest that F-CH2O formation is favorable in both kinetics and thermodynamics,which is consistent with the previous report[36].Except for hydrogenation,the CHO group may also undergo the C-O bond scission (R5).However,this process should be insignificant,as it needs to overcome energy barrier as high as 1.77 eV,with reaction energy of 0.53 eV.In TS5,the C-O bond is increased to 2.105 Å.
Further hydrogenation of F-CH2O to form FCH2OH requires an energy barrier of 1.12 eV.In addition,the F-CH2OH can be also fabricated through hydrogenation of F-CHOH,with the energy barrier of 0.35 eV.In TS6 and TS7,the O7-H and C6-H bond lengths are decreased to 1.575 and 1.924 Å,respectively.Based on the above calculated results,furfuryl alcohol is more favorable to be produced through the F-CH2O intermediate (black line) via the route of F-CHO+2H→F-CH2O+H→F-CH2OH with the highest energy surface of 0.58 eV,rather than through F-CHOH intermediate (red line).In the favorable pathway,F-CH2O+H→F-CH2OH is the ratelimiting step,requiring the activation energy of 1.12 eV.
2.3.2 Mechanism on Cu(211) surface
The energy profiles and the optimized structure for the furfuryl alcohol formation on the Cu(211)surface were provided in Figure 5.
The F-CHO+H was adopted as the initial state,and the hydrogenation of C6and O7of F-CHO to form F-CH2O and F-CHOH were then considered,respectively.The activation energy barriers and corresponding reaction energies of these two steps are 0.52 and -0.15 eV,and 1.08 and 0.49 eV,respectively.In TS3*,H is adsorbed on the top site of Cu atom,with the H-C6bond length of 1.679 Å.In TS4*,the H-O7bond length is shortened from 3.753 Å of the initial state to 1.372 Å of optimized state.Similar to the results on Cu(111) surface,the detachment of O atom from CHO group of F-CHO is impossible,as the energy barrier reaches 1.75 eV,with reaction energy of 0.98 eV.In TS5*,the C-O bond length is 1.950 Å.When the F-CH2O+H and FCHOH+H were used as the initial states,the formation of furfuryl alcohol requires activation energies of 0.78 and 0.48 eV,with the reaction energies of -0.09 and -0.73 eV,respectively.In TS6*and TS7*,the O7-H and C6-H bond lengths are shortened from the initial 3.663 and 3.346 Å to 1.501 and 1.869 Å,respectively.Therefore,F-CHO +2H→F-CH2O+H→F-CH2OH is also the dominant pathway for furfuryl alcohol formation on Cu(211)surface,and the highest energy surface of the whole reaction is 0.63 eV.Meanwhile,F-CH2O+H→FCH2OH is the rate-limiting step,requiring the activation barrier of 0.78 eV.
2.3.3 Comparison of mechanism between Cu(111)and Cu(211) surface
In order to evaluate effect of Cu catalyst structure on the kinetics in furfuryl alcohol formation,the optimal pathway (e.g.F-CHO+2H→F-CH2O +H→F-CH2OH) on these two surfaces were compared,as depicted in Figure 6.On Cu(111) surface,the first and second hydrogenations of F-CHO to form furfuryl alcohol need to overcome energy barriers of 0.54 and 1.12 eV on Cu(111) surface (black line),being higher than those on Cu(211) surface (0.52 and 0.78 eV) (red line).This confirms that Cu(211) surface should have higher reactivity to furfuryl alcohol formation.
2.4 Effect of Cu catalyst structure for furfuryl alcohol formation
To get further insight into the effect of electronic structure of Cu(111) and Cu(211) surface,the charge density difference of furfural adsorption was calculated.As shown in Figure 7(a),the charge density accumulation suggests that furfural mainly interacts with Cu(111) surface via O7atom of aldehyde group (CHO),due to low affinity between C and Cu atoms,whereas for Cu(211) surface,the atom of O7of CHO group and the C3,C4and C5atoms of the furanic ring can interact with the step edge of Cu(Figure 7(b)).It is worthy to note that the electron density accumulation and consumption on Cu(211)surface are larger than that on Cu(111) surface.To better understand the charge transfer from F-CHO to Cu(111) surface or Cu(211) surface,the Bader charge population analysis was further carried out and it gave the similar results as that of charge density difference analysis.In addition,thedproject density of states of Cu(111) and Cu(211) surface was calculated,to reveal the interaction between the adsorbates and the surface metald-band[22,42].As shown in Figure 7(c) and 7(d),thed-band centers of Cu(111) and Cu(211) are -2.40 and -2.24 eV,respectively.This also demonstrates the stronger interaction between the adsorbate and the Cu(211)surface,as thed-band center of Cu(211) surface is closer to Fermi level[43,44].On the other hand,the geometric effect may play a key role in furfural adsorption on Cu(111) and Cu(211) surface.The bond length between O7and Cu on Cu(211) surface(dO7-Cu=1.997 Å) is shorter than that on Cu(111)surface (dO7-Cu=2.105 Å).Both the electronic and geometric effects result in the stronger adsorption of F-CHO on Cu(211) (Eads=-1.43 eV) than that on Cu(111) surface (Eads=-0.70 eV). Therefore,introduction of step active sites into Cu catalyst promotes the conversion of furfural to furfuryl alcohol.

Figure 6 Most favorable pathway for furfuryl alcohol formation on Cu(111) surface and Cu(211) surface
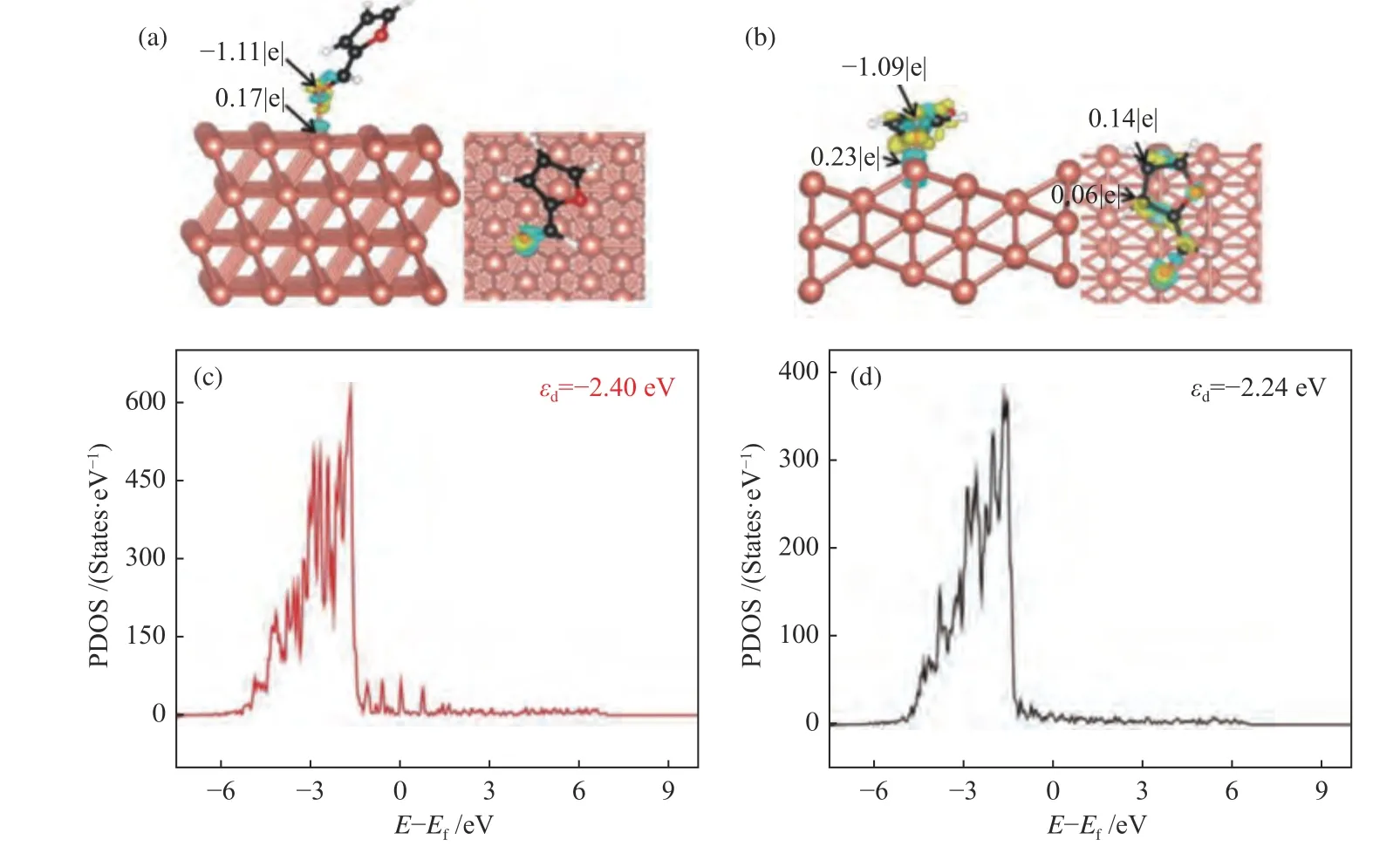
Figure 7 Differential charge diagram (a) and Bader charge (b) of furfural adsorption on Cu(111) and Cu(211) surface;d-projected density of states (PDOS) of the Cu atoms on Cu(111) (c) and Cu(211) (d) surfaces
3 Conclusions
In summary,hydrogenation of furfural into furfuryl alcohol on Cu(111) surface and Cu(211)surface was investigated by DFT calculations,to unravel the structure sensitivity of Cu catalyst for furfural conversion.The calculated results indicate that F-CH2O is the dominant intermediate for furfuryl alcohol formation on Cu(111) and Cu(211) surfaces,along with the F-CH2O+H→F-CH2OH serving as the rate-limiting step.Stepped Cu(211) surface is predicted to have a higher reactivity than Cu(111) surface.This is because of: (i) the promotion of the dissociation of H2;(ii) the enhancement of the adsorption of furfural,which is confirmed by the results of charge density difference andd-band center.Therefore,controlling the catalyst structure can effectively improve the catalytic performance of furfural hydrogenation.The insight shown in this work provides a new horizon for practical catalytic processes involved in the refining of biomass-derived oils.
