
机械球磨法制备NiMo 催化剂及其在菲加氢中的应用
2023-02-07 08:00王斐钟梅李建亚力昆江吐尔逊靳立军
燃料化学学报 2023年2期
王斐,钟梅,*,李建,亚力昆江·吐尔逊,靳立军,2,*
(1. 新疆大学化工学院 省部共建碳基能源资源化学与利用国家重点实验室 新疆煤炭清洁转化与化工过程重点实验室,新疆 乌鲁木齐 830046;2. 大连理工大学化工学院 精细化工国家重点实验室 煤化工研究所 辽宁 大连 116024)
中低温煤焦油作为低阶煤600-800 ℃热解的液体产品,其多环芳烃含量丰富,目前,主要用于加氢生产汽柴油,这导致产品种类单一、附加值低。因此,如何实现其多元化、高值化与精细化利用,是煤化工行业亟需解决的关键问题。由于此类焦油中2-3 环的芳烃比例较高[1,2],通过控制加氢深度来制取高密度燃料[3],不仅可以实现中国煤焦油资源的高价值利用,还能保障中国航空航天事业的发展。
芳烃可控加氢的关键在于构建高活性的催化剂,主要包括贵金属类[4]、非贵金属类[5,6]。贵金属催化剂的加氢活性高,但价格昂贵,抗硫氮性能差[7]。非贵金属单质Ni 具有较好的H2解离能力[5],常用作加氢催化剂的活性组分,特别是引入P 形成金属磷化物,其加氢活性可与贵金属活性相当[8],然而此类催化剂的晶相易发生相变,导致催化剂失活[9]。相对而言,硫化态的NiMo 催化剂不仅活性高,且价格低廉,受到广泛关注。Zhang 等[10]采用共沉淀法制备NiMo/Al2O3催化剂,考察焙烧温度对菲加氢性能的影响。结果表明,随着煅烧温度的升高,Ni 和Al2O3相互作用增强,非活性的NiAl2O4增加,活性组分中的Ni/Mo 比下降,导致催化活性降低,当温度达800 ℃时,菲的转化率降低至62%。Jeong 等[11]采用水热合成法制得Ni 掺杂的MoS2催化剂,发现Mo 的硫化程度随Ni/Mo比增加先提高后降低,当Ni/Mo 比为0.45 时达到最高,Ni 多以NiMoS 相存在。该催化剂在重油加氢过程中油产率与Mo 的硫化程度变化呈现出相同的趋势,最大油产率80.5%,较未掺杂Ni 时高16.4%,固体残渣则降低至5.4%。Zhang 等[12]对比不同方法制得的Al2O3负载Ni、Mo 活性金属的复合催化剂结构特征,研究显示,与拟薄水铝石直接焙烧制得的Al2O3载体相比,以Al(NO3)3为原料通过固相研磨法制得Al2O3为载体制备的催化剂具有独特的双峰多孔结构,不仅活性位点丰富,而且有利扩散,二苯并噻吩转化率更高。Qiu 等[13]发现,相较于等体积浸渍法,顺序浸渍法制得催化剂的活性组分分散性更高、酸性更强。特别是Ni 负载量为6.25%、Ni/(Ni+Mo) 比为0.4 时,其作用下喹啉的脱氮率可达71.7%。虽然上述催化剂加氢活性较高,但制备过程复杂、耗时长,活性金属易在载体表面聚集。本课题组前期研究表明,机械球磨法制备催化剂具有过程简单、活性组分分散均匀、易于放大、催化剂活性高等特点[14-16],因此,本论文采用机械球磨法制备介孔NiMo 催化剂,探究不同Ni/(Ni+Mo)比及恒定Ni/(Ni+Mo)比时Ni 和Mo 的含量以及硫化剂硫代硫酸铵(ATS) 的负载量对焦油模型化合物菲加氢产物分布的影响,阐明催化剂组成和结构对目标产物选择性的调控规律。
1 实验部分
1.1 试剂
实验所用药品均为分析纯,其中,Al(NO3)3·9H2O购自于天津市致远化学试剂有限公司,(NH4)2CO3购自于天津市盛淼精细化工公司,Ni(NO3)2·9H2O购自于上海山浦化工有限公司,(NH4)6Mo7O24·4H2O、菲(97%)、硫代硫酸铵(99%)、正十二烷(98%)均购自于上海麦克林生化科技有限公司,正庚烷购自于天津市北联精细化学品开发有限公司。
1.2 催化剂的制备
称取一定量的Al(NO3)3·9H2O、(NH4)6Mo7O24·4H2O、Ni(NO3)2·9H2O 和(NH4)2CO3置于球磨罐中,球磨1 h、干燥12 h 后,于500 ℃焙烧3 h 制得NiMo/Al2O3,记为NixMoy-z,其中,x和y分别为Ni 和Mo在催化剂中的质量含量,z为Ni/(Ni+Mo)物质的量之比。
然后按照S/Mo 物质的量之比为3.0 和4.5,称取一定量硫代硫酸铵(ATS),采用等体积浸渍法将其浸渍于NiMo/Al2O3上,室温下老化12 h 后于90 ℃干燥3 h,制得硫化态的NiMo 催化剂,记为NixMoyz-φ,φ为预硫化时的S/Mo 比。为对比,采用相同的方法制得氧化铝载体。
1.3 催化性能评价
将0.1 g 催化剂置于100 mL 高压釜中进行预硫化,设置氢气初压为3 MPa,升温速率为4.5 ℃/min,由室温升至300 ℃并保持60 min。待降至室温后,加入含1%菲(PHE)的正庚烷溶液20 g,经氮气吹扫三次后,将氢气压力升至5 MPa,在400 r/min转速下,以10 ℃/min 将温度升至340 ℃,并保持4 h。为了分析菲的加氢路径,在相同的实验条件下,反应20 min 后(240 ℃),每5 min 取一次样,直至340 ℃,随后取样时间间隔为30 min,总时长设定为240 min。
1.4 催化剂的表征
对硫化后的催化剂进行结构表征。其中,晶体结构由德国布鲁克AXS 有限公司的GmbH/D8 Advance 型多晶粉末衍射仪测定,10°-90°扫描。采用美国 Micromeritics ASAP 2020 物理吸附仪测定催化剂的孔道结构,脱附温度为150 ℃。在法国HORIBA Scientific 公司的Horiba Evolution 型激光拉曼光谱仪上测定样品的拉曼光谱特征,激光器波长为532 nm,记录波长为200-1200 cm-1。元素的形态分布由美国Thermo Fisher Scientific 的ESCA Lab 250Xi 电子能谱仪测定,通过Avantage 软件对XPS 光谱进行分析。采用日本电子JEM-2100高倍透射电镜观察催化剂的形貌,其加速电压为300 kV。
1.5 液体产物分析及转化率、选择性计算
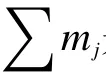
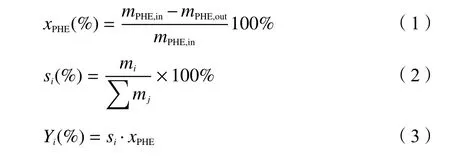
式中,xPHE—菲的转化率;mPHE,in—反应初始菲的质量;mPHE,out—反应终止菲的质量;s∑i—加氢产物i的选择性;mi—加氢产物i的质量;是所有加氢产物总质量;Yi—i物质的收率。
2 结果与讨论
2.1 催化剂表征与分析
采用XRD 对NiMo 催化剂进行结构表征,由图1(a) 可以看出,在37.5°、45.8°和66.8°处出现了Al2O3的特征衍射峰(JCPDS 10-0457)[17],其峰型较宽意味着Al2O3的结晶度较低[18]。当Ni/(Ni+Mo)比≤0.33 时,未检测到Ni 物种的衍射峰,说明其分散性较好,当进一步提高Ni/(Ni+Mo) 比时,在18.6°处出现了新的衍射峰,意味着Ni3S2、Ni9S8或Ni3S4的出现[19],这是由于焙烧后部分Ni 未掺杂在氧化钼中或与载体结合,而是以NiO 的形式存在,硫化后形成了非活性的NixSy[20]。从图1(b) 可见,当固定Ni/(Ni+Mo) 比为0.33,进一步提高Mo 和Ni 的含量分别至20%和6%时,仍未观察到明显的Ni、Mo 物种的衍射峰,这可能是由于在机械球磨过程中,强烈的机械碰撞和剪切作用使得各活性组分均匀分散[21]。此外,提高硫代硫酸铵(ATS)的量,将S/Mo 比由3.0(Ni4.8Mo16-0.33-3)增加至4.5 时,Ni4.8Mo16-0.33-4.5 催化剂上也无硫化钼或硫化镍的特征衍射峰,这意味着硫化未改变活性组分的晶型结构。
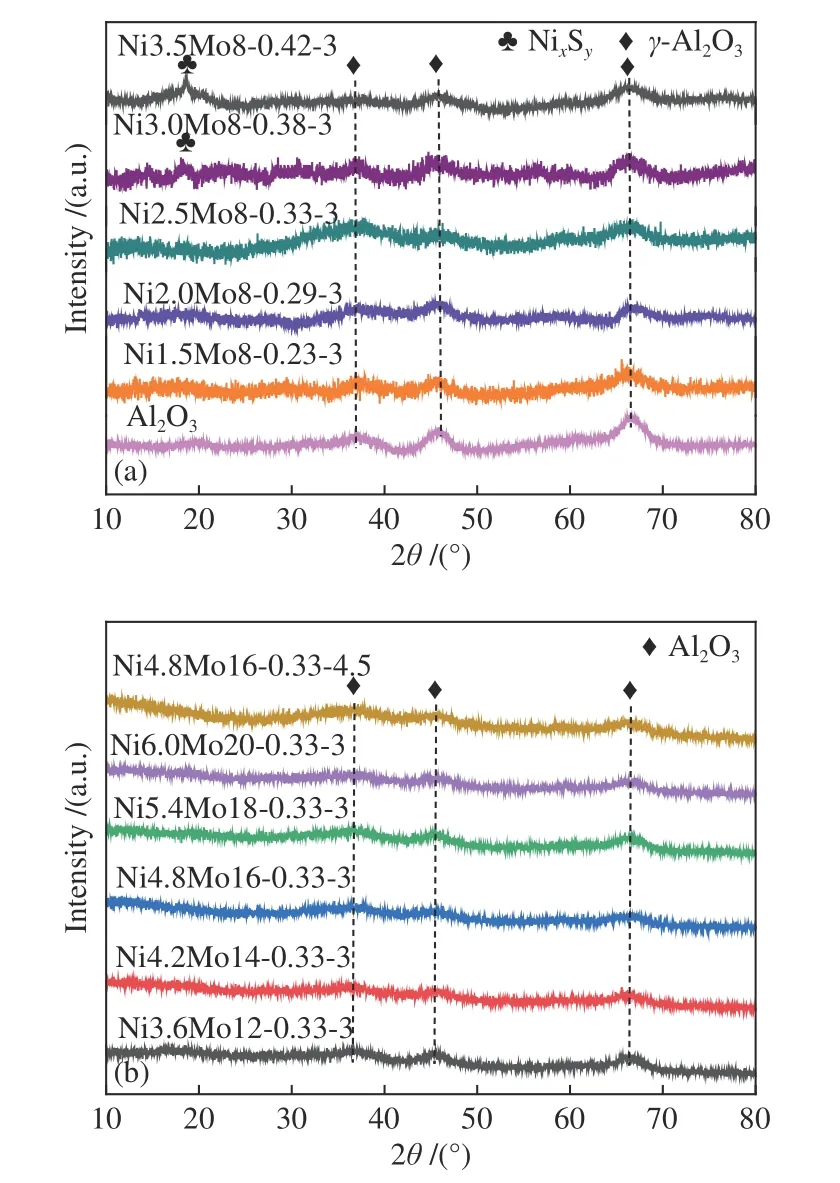
图1 不同催化剂的XRD 谱图Figure 1 XRD patterns of catalysts: (a) different Ni/(Ni+Mo)ratios;(b) constant Ni/(Ni+Mo) ratio and different S/Mo ratios
采用TEM-mapping 对Ni、Mo、S 的分布进行分析,如图2 所示。Ni、Mo、S 元素均匀分布于氧化铝载体中,但当Ni/(Ni+Mo) 比超过0.33 时,Ni元素的高强度位置与S 元素存在差异,这可能是由于在含镍量较高的样品中,部分Ni 与S 形成NixSy高亮团簇[11],与XRD 谱图结果一致。
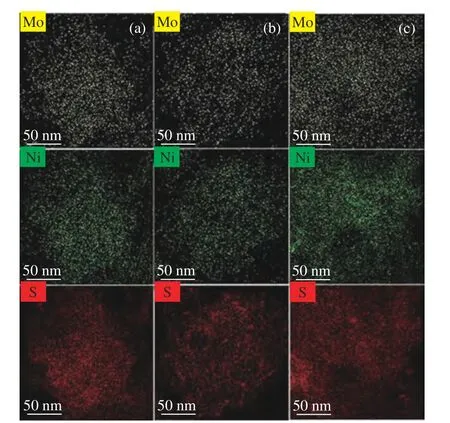
图2 元素分布图Figure 2 Elemental mapping images: (a) Ni1.5Mo8-0.23-3;(b) Ni2.5Mo8-0.33-3;(c) Ni3.5Mo8-0.42-3
由图3 催化剂的氮气吸附-脱附等温曲线可知,所有的等温线均为IV 型,具有H2 型回滞环[22],表明催化剂是典型的介孔材料。在相对压力(p/p0)为0.4-0.6 时,曲线非常陡峭,这归因于氮气在介孔中的毛细管冷凝现象[23]。在较高的相对压力(p/p0>0.8)下,曲线接近于水平,表明无大孔形成[24]。图3(c)和图3(d)显示,孔径分布为2-10 nm,平均孔径为3-5 nm。当Ni/(Ni+Mo) 比由0.23 增加至0.42 时,比表面积和孔体积先增加后降低,于0.33 处最大。分析认为,Ni 促进了Mo 的分散,而非仅沉积于孔道中。Ni/(Ni+Mo) 比超过0.33 时,过多的Ni团聚并堵塞孔道,硫化后形成硫化镍团簇。当恒定Ni/(Ni+Mo)比为0.33,Ni 和Mo 含量增加时,堵孔效应愈发明显,致使催化剂的比表面积和孔径显著减小(表1)。另外,当硫代硫酸铵(ATS)的质量增加时,催化剂的比表面积和孔体积都随之增加,这是预硫化过程中ATS 分解产生气体的二次扩孔所致[25]。
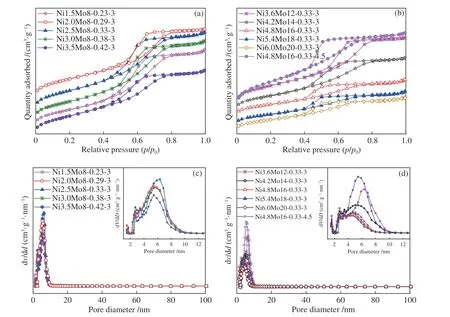
图3 催化剂的N2 吸附-脱附等温曲线和孔径分布Figure 3 N2 adsorption-desorption isotherms ((a),(b)) and pore size distributions of catalysts ((c),(d)): ((a),(c)) different Ni/(Ni+Mo) ratios;((b),(d)) constant Ni/(Ni+Mo) ratio and different S/Mo ratios
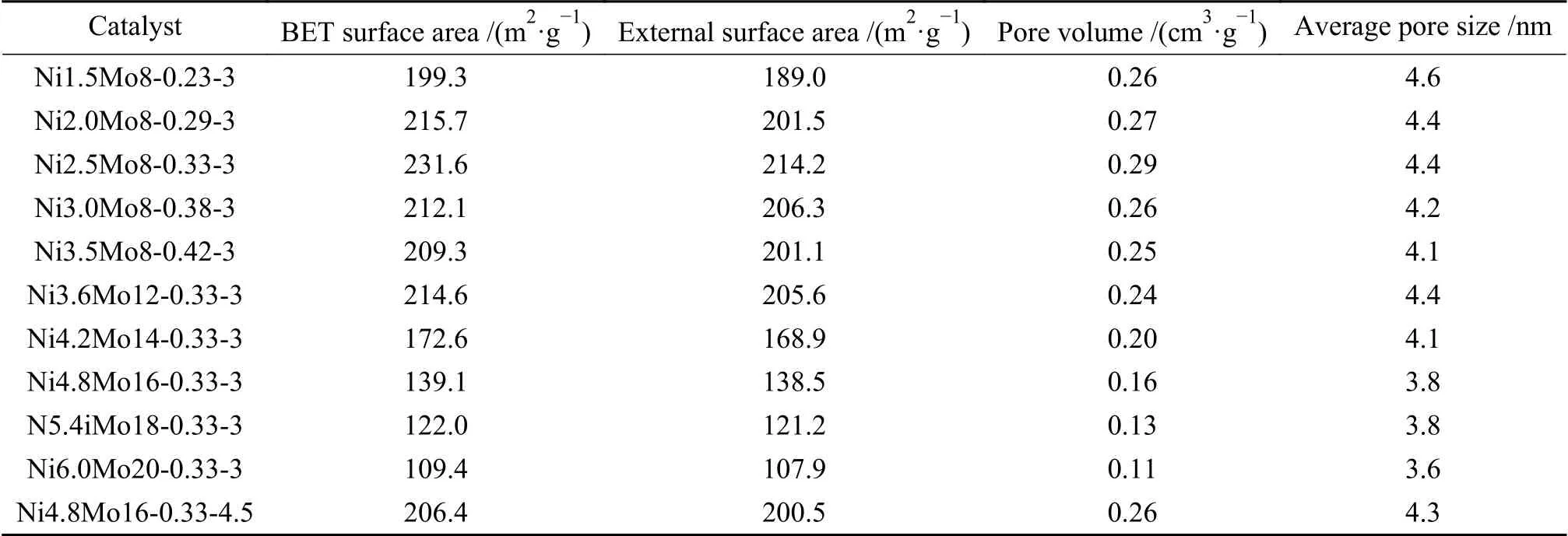
表1 催化剂的孔结构特征Table 1 Pore structure characteristics of catalysts
图4 为催化剂的拉曼光谱谱图。由图4 可知,分别在405 和380 cm-1处附近,各催化剂均出现了特征峰,这可归属于MoS2的面外振动模式(A1g)和面内振动模式(E12g)。由于A1g和E12g之间的距离Δω与MoS2堆砌层数正相关[26],A1g峰蓝移、E12g峰红移,故Ni/(Ni+Mo)比增加时Δω值减小,说明MoS2堆砌层数减小。意味着适量Ni 促进了Mo的分散,有利于形成堆积层数少的MoS2。然而,进一步增加Ni/(Ni+Mo) 比至0.42 时,Δω反而增加,可归因于NixSy簇的形成。恒定Ni/(Ni+Mo)比,随着Mo 的硫化程度增加(表2),Δω并不改变,但A1g峰发生蓝移,表明Ni 是以n型掺杂于MoS2中,形成的NiMoS 活性相是缺电子结构[27],有利于反应物吸附。当增加S/Mo 比为4.5 时,仅A1g峰发生蓝移,Δω值仍不变,意味着MoS2的堆积层数未发生变化。

表2 Mo、Ni 和S 不同形态的原子比Table 2 Atoms percentage in different Mo,Ni and S forms (%)
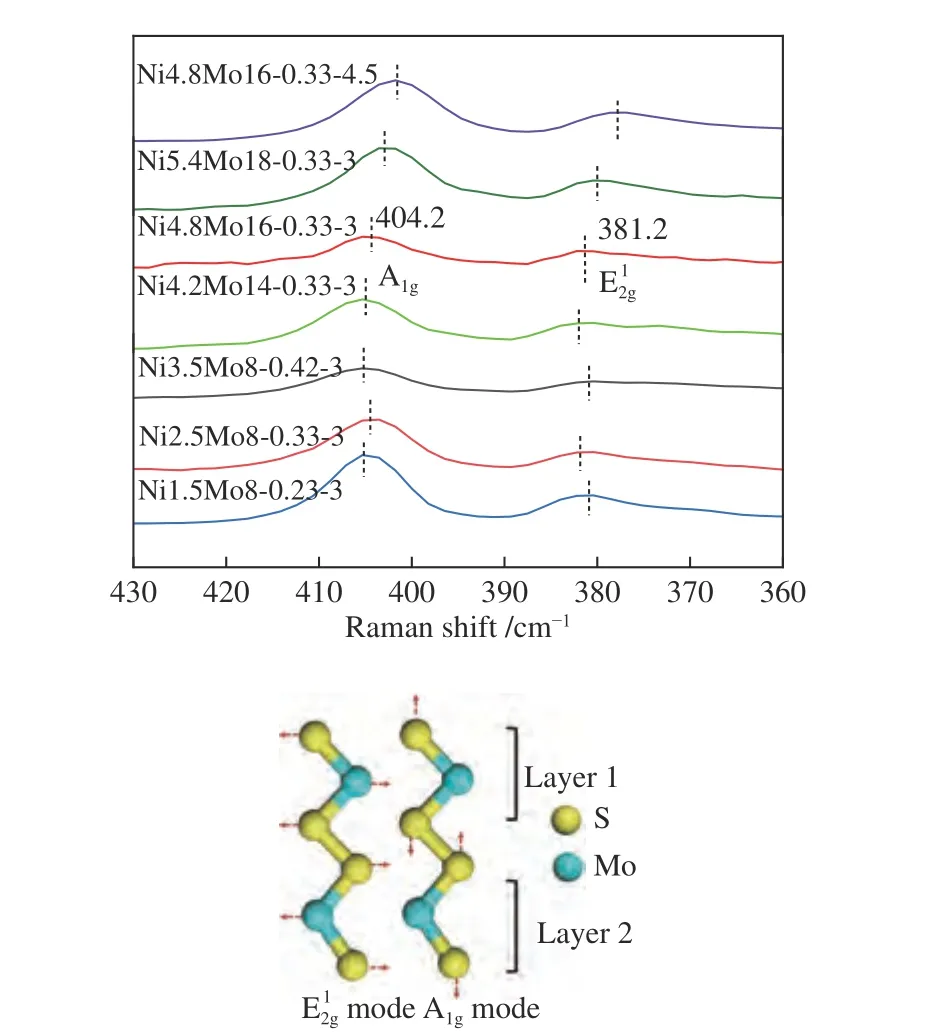
图4 催化剂的拉曼光谱谱图Figure 4 Raman spectra of catalysts
图5 给出了不同Ni/(Ni+Mo)比催化剂的晶格结构及堆积层数分布。由图5 可以看出,各催化剂表面均存在明显的晶格条纹。当Ni/(Ni+Mo)比≤0.33 时,仅观测到MoS2的晶格条纹,晶面间距为0.61 nm。当该比值由0.23 增加至0.33 时,MoS2的堆积层数分布的峰值左移(图5(e)),计算得到平均堆积层数由5.1 下降至4.2。这是由于适量Ni掺入MoS2中导致MoS2层间的交联减弱[28],有利于更多的活性位点暴露。当Ni/(Ni+Mo)进一步提升至0.42 时(图5(c)),出现了间距为0.22 nm 的NixSy晶格条纹[29],致使MoS2的平均堆积层数增至4.6。恒定Ni/(Ni+Mo) 比为0.33,Ni4.8Mo16-0.33-3 催化剂中MoS2的堆积层数为4.3,与Ni2.5Mo8-0.33-3的堆积层数相近。
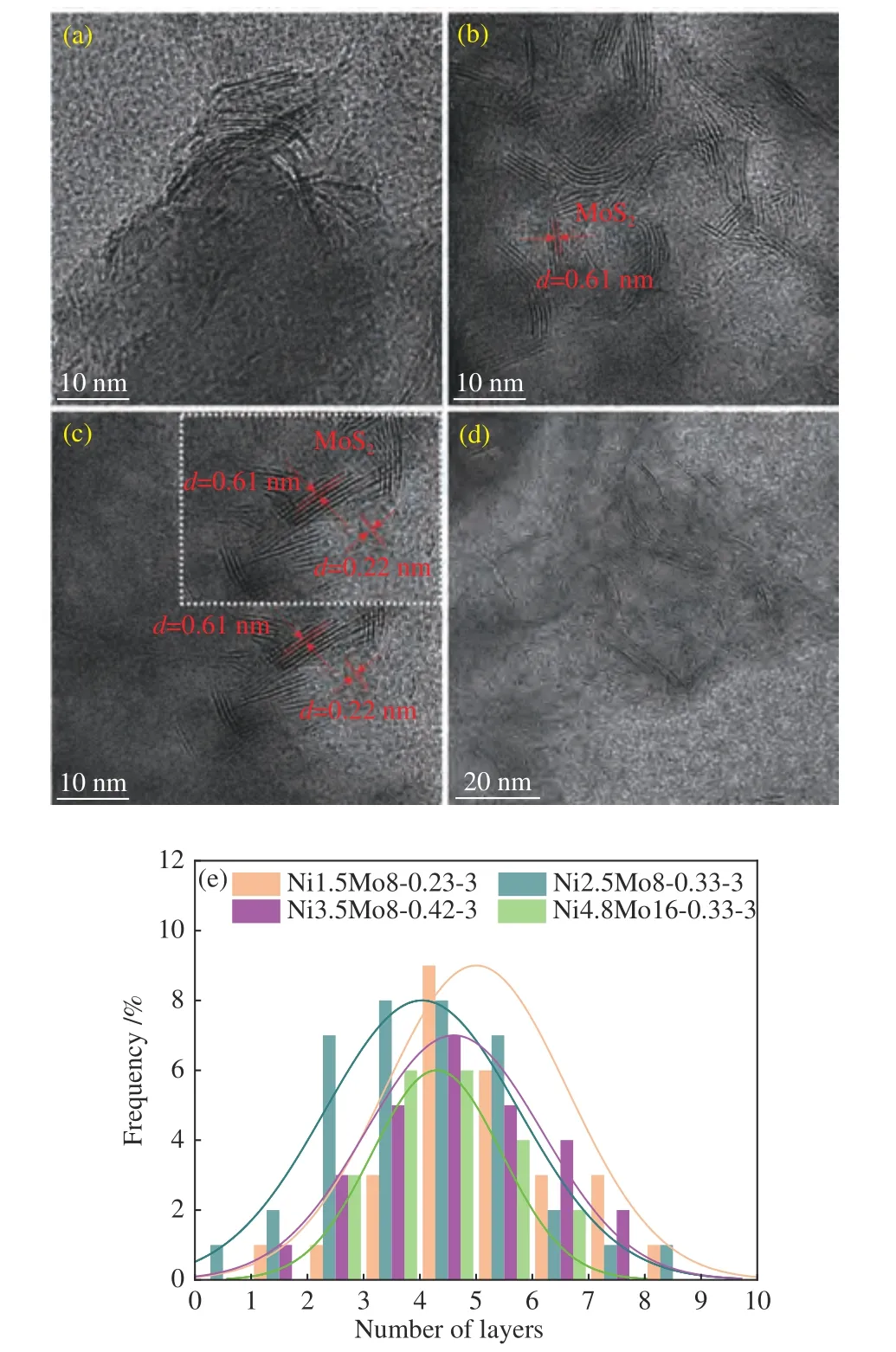
图5 催化剂的HRTEM 图及堆积层数分布Figure 5 HRTEM images and accumulation layer number distribution of catalysts: (a) Ni1.5Mo8-0.23-3;(b) Ni2.5Mo8-0.33-3;(c) Ni3.5Mo8-0.42-3;(d) Ni4.8Mo16-0.33-3;(e) accumulation layer number distribution
为了获得催化剂中不同金属物种相对含量、化学状态和金属物种硫化程度,对硫化后的NiMo催化剂进行了XPS 分析,如图6 所示。从XPS 全谱谱图(图6(a))可以看出,催化剂中存在Mo 3d(229.1 eV)、S 2p(163.1 eV)、Ni 2p(855.4 eV)和Al 2p(74.9 eV)的特征峰。图6(b)中,228.8、232.3 eV 分别代表Mo 3d5/2和Mo 3d3/2MoS2的特征峰(MoIV);位于232.7、235.8 eV 处的特征峰分别归属于Mo 3d5/2、Mo 3d3/2氧化钼的峰(MoVI);在230.70、233.8 eV 处的特征峰分别属于MoOxSy相(MoV)的Mo 3d5/2和Mo 3d3/2的峰[30]。随着Ni/(Ni+Mo)比增加,MoVI的含量随之降低,MoIV的含量先增加后降低(表2),于Ni/(Ni+Mo)比0.33 时达最高值31.7%。根据文献可知,MoIV的含量代表Mo 的硫化程度[31],意味着该比值下Mo 的硫化程度最高。进一步提高Ni/(Ni+Mo)比,过量Ni 形成NixSy簇,导致Ni 促进Mo 的硫化效应降低,更多的Mo 与S 和O 相互结合生成MoOxSy。恒定Ni/(Ni+Mo)比,增加Ni 和Mo 的含量时,活性金属与载体的相互作用减弱[32],这有利于Mo 的硫化,因而MoIV的相对含量增加。提高固态硫化剂ATS 的量使得S/Mo 比增至4.5,意味着硫源增加,因而MoIV含量显著增加,达到62.8%。
图6(d) 是Ni 2p3/2的XPS 能谱图。Ni 掺杂在MoS2中形成的NiMoS 相一般在853.5 eV 处出现,但因机械球磨法过程中会使各个组分产生一定的相互作用,致使Ni 的电子云密度偏低[35],因而Ni的结合能偏移至854.3 eV 处。NixSy的峰出现在852.3 eV 处,856.5 eV 的特征峰属于Ni 与载体形成弱相互作用的氧化镍(Ni2+a),859.5 eV 处的特征峰则为强相互作用形成的镍铝尖晶石(Ni2+b),862.8 eV 处是Ni 的卫星峰[36]。当Ni/(Ni+Mo)比由0.23 增至0.42 时,虽然催化剂中Ni 的含量增加,但NiMoS 含量由57.7%降至20.3%(表2),体现镍与载体强相互作用的Ni2+b则从10.1%增至50.1%(表2),说明Ni 虽能与MoⅣ形成NiMoS,但生成量有限,故当其含量增加后,更多的Ni 融入到Al2O3中形成镍铝尖晶石[37],或形成NixSy沉积于孔道内,致使催化剂的比表面积降低(表1)。恒定Ni/(Ni+Mo)比为0.33,NiMoS 的相对含量持续增加。增加固态硫化剂ATS 的量(Ni4.8Mo16-0.33-4.5)进一步提高了Mo 的硫化程度,NiMoS 相含量提高至50.8%。
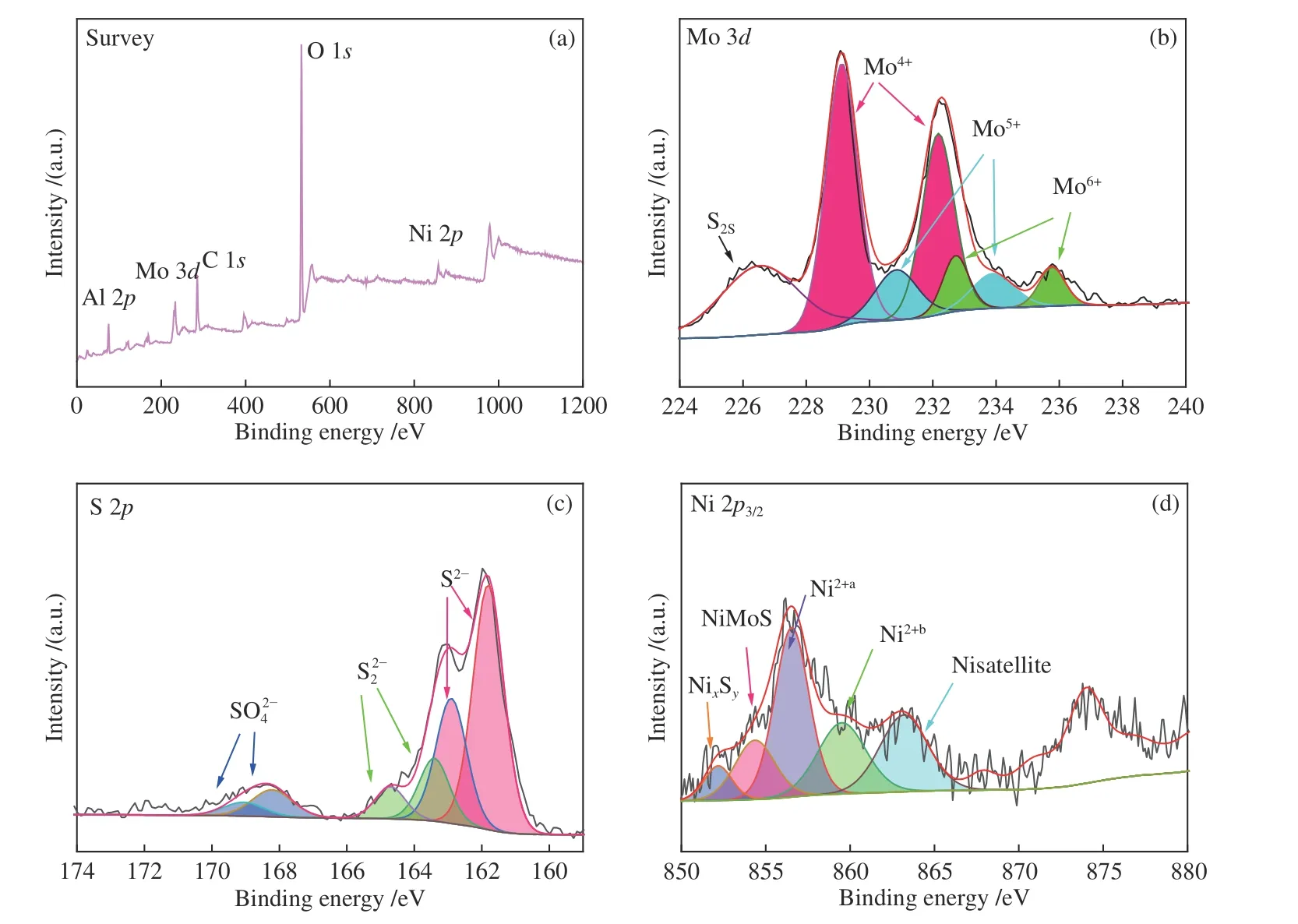
图6 XPS 分峰拟合图Figure 6 Curve-fitting of the XPS spectra: (a) full spectrum;(b) Mo 3d;(c) S 2p;(d) Ni 2p3/2
2.2 菲加氢性能评价
图7 给出了在催化剂作用下焦油模型化合物菲的转化率与产物的选择性。由图7(a)可知,随着Ni/(Ni+Mo) 比增加,菲的转化率先增加后降低。当该比值为0.33 时(Ni2.5Mo8-0.33-3),菲的转化率达到最大值74.7%,产物中9,10-二氢菲(DHP)和1,2,3,4-四氢菲(THP) 的选择性均最低,八氢菲(OHP)的选择性最高,特别是1,2,3,4,5,6,7,8-八氢菲(sym-OHP) 的选择性明显高于其他物质。然而,进一步增加Ni/(Ni+Mo)比,不仅菲的转化率下降,产物分布变化亦很明显。其中,DHP 和THP的选择性显著提高,而OHP 和全氢菲(PHP)的选择性降低,这与表1 和表2 中催化剂的比表面积和MoⅣ的变化趋势一致。这是因为Ni/(Ni+Mo)比适量增加时,Ni 促进了Mo 的分散,有利于形成更多的NiMoS 活性相,提高芳烃π电子向金属d轨道转移的能力[38],同时MoS2的堆积层数降低使得更多的活性位点被暴露出,这于菲的转化有利。然而该比值过高时,过量的Ni 形成NixSy团簇沉积于孔道,导致催化剂的比表面积降低,反而抑制了Mo 的硫化(表2),形成了更多低活性的MoOxSy,或镍铝尖晶石,从而抑制菲的转化和深度加氢。
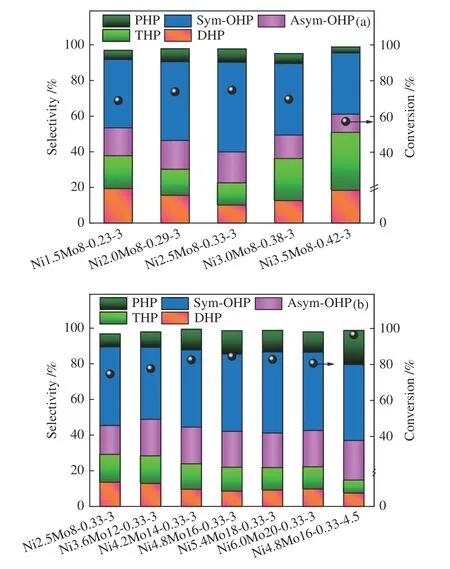
图7 菲的转化率及液体产物分布Figure 7 Conversion of phenanthrene and distribution of liquid products: (a) different Ni/(Ni+Mo) ratios;(b) constant Ni/(Ni+Mo) ratio and different S/Mo ratios
恒定Ni/(Ni+Mo)比为0.33,同时提高Ni 和Mo的含量菲的转化率先升高后降低,当Ni 和Mo 含量分别为4.8% 和16% 时(Ni4.8Mo16-0.33-3),菲的转化率达到最大值84.5%。此时OHP 和PHP 的选择性均最高(图7(b))。这是由于Ni 和Mo 含量增加时,催化剂的比表面积急剧降低(表1),导致两种金属组分的分散性下降,暴露的活性位点减少,这于菲的加氢不利;另一方面,Mo 的硫化程度持续增加(表2),这促进了菲的转化。两种效应相互竞争,当Ni 和Mo 的含量超过一定值时,前者的作用更为显著。与Ni4.8Mo16-0.33-3 相比,S/Mo 比增至4.5(Ni4.8Mo16-0.33-4.5)提高了Mo 的硫化程度(表2)和催化剂的比表面积(表1),因而催化剂的加氢活性增强,相应的,菲的转化率增至96.5%,八氢菲和全氢菲的总选择性和产率分别提高至83.9%、80.9%。由此可见,提高催化剂的比表面积和Mo 的硫化程度可以提高催化剂的反应活性和产物选择性,实现菲的定向转化生成八氢菲。
2.3 菲加氢反应路径
探究了Ni4.8Mo16-0.33-4.5 作用下菲加氢过程的反应路径,对不同反应时间产物进行分析。结果如图8 所示。由图8(a) 可以看出,反应时间为20 min 时,四氢菲THP 的选择性最高,其次为DHP,这是由于菲的基态最高活性碳位在C9 和C10[39,40],第二高活性碳位是C1,两处均易吸附于活性位点形成π-σ*键,进而生成DHP 和THP,且DHP 可进一步通过加氢转移生成THP 和1,2,3,4,4a,9,10,10a-OHP(asym-OHP),THP 则继续加氢生成sym-OHP。因此,随反应时间的延长,DHP 和THP 的选择性持续下降,而菲的转化率增加,当两者的生成速率高于转化速率时,收率增加,反之则减小。反应150 min 后,DHP 和THP 的收率降低(图8(b)),说明菲以深度加氢生成八氢菲和全氢菲为主,首先生成DHP 和THP 的选择性持续降低。此外,THP 的选择性始终高于DHP,这是由于机械球磨过程中金属镍与载体的相互作用促进了电子转移,形成具有Ni 缺电子结构的NiMoS 活性相,有利于芳烃中的π电子向金属d轨道转移形成σ键[38],从而促进菲从侧环加氢,THP 则继续加氢生成OHP。基于上述分析,菲加氢路径如图9所示。
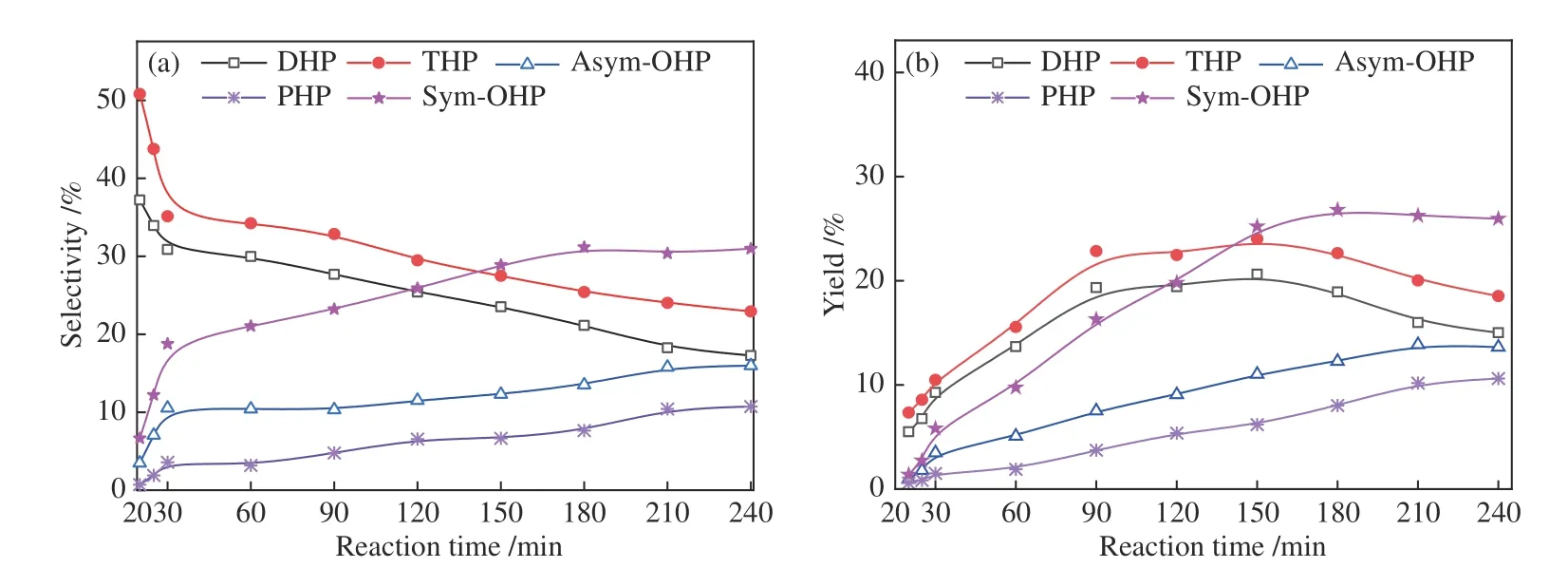
图8 Ni4.8Mo16-0.33-4.5 作用下菲加氢产物选择性和收率随时间的变化Figure 8 Selectivity and yield of phenanthrene hydrogenation products under Ni4.8Mo16-0.33-4.5 reaction with time

图9 Ni4.8Mo16-0.33-4.5 作用下菲的加氢反应路径示意图Figure 9 Phenanthrene hydrogenation reaction path under the action of Ni4.8Mo16-0.33-4.5
3 结论
本研究采用机械球磨法制备了不同NiMo 含量的双金属硫化态催化剂,探究了Ni/(Ni+Mo)比、恒定Ni/(Ni+Mo)比时Ni 和Mo 的含量以及硫化剂硫代硫酸铵(ATS)的负载量对焦油模型化合物菲加氢产物分布的影响,结论如下。
采用球磨法制备的介孔催化剂,孔径集中于2-10 nm,Ni 和Mo 分散均匀。Ni/(Ni+Mo)原子比显著影响活性组分粒径和表面性质。适量Ni的加入不仅能够降低MoS2的堆积层数,还能促进Mo 硫化形成更多的活性MoⅣ。Ni/(Ni+Mo) 比超过0.33 时,过量的Ni 会以NixSy团簇的形式覆盖活性位点,导致催化剂的比表面积和孔容降低,不利于催化反应进行。
恒定Ni/(Ni+Mo)原子比为0.33,提高Ni 和Mo含量时,NiMoS 活性相增加,比表面积和孔容下降;增加S/Mo 比,催化剂的比表面积和MoIV的含量均增加。
随着Ni/(Ni+Mo) 比的增加,菲的转化率先上升后下降,在Ni2.5Mo8-0.33-3 作用下达到最大值74.7%。恒定Ni/(Ni+Mo)比为0.33,当Ni 和Mo 的含量分别为4.8%和16%时,菲的转化率为84.5%,进一步提高S/Mo 比至4.5,菲的转化率增至96.5%,八氢菲和全氢菲的总选择性和收率分别达83.9%和80.9%。当Ni 和Mo 的含量低于4.8% 和16%时,Mo 的硫化程度对菲加氢行为的影响占主导地位,之后比表面积的作用效果更为显著。在菲的深度加氢过程中,主要由四氢菲从侧环加氢生成全氢菲。
猜你喜欢
机械工业标准化与质量(2022年6期)2022-08-12
科技创新与应用(2020年19期)2020-06-23
小资CHIC!ELEGANCE(2019年40期)2019-12-10
中国特种设备安全(2019年3期)2019-04-22
中国调味品(2017年2期)2017-03-20
山东工业技术(2016年15期)2016-12-01
中国水能及电气化(2016年11期)2016-02-28
环境科技(2015年5期)2015-11-08
中学化学(2015年2期)2015-06-05
理科考试研究·高中(2014年8期)2014-10-17

- 燃料化学学报的其它文章
- 生物质“热溶富碳”及其产物利用
- 麦秸秆与黑龙江褐煤共热氧化法制备腐植酸及其结构分析
- 费托合成铁基催化剂中Fe3O4 含量对CO2 选择性的影响
- 引入Al2O3 对Pt/ZSM-23 催化剂加氢异构性能的调控
- Cerium-modified copper/hexagonal mesoporous silica catalyst for efficient dimethyl oxalate hydrogenation to ethylene glycol under moderate reaction conditions
- Nanosized amorphous nickel-boron alloy electrocatalysts for hydrogen evolution reaction under alkaline conditions