
量子化学研究不同取代基对六苯并蔻电荷传输性能的影响
2023-02-04 03:18陈自然张宇红何展荣余文浩
云南大学学报(自然科学版) 2023年1期
陈自然,张宇红,李 渊,何展荣,余文浩
(1.四川职业技术学院 建筑与环境工程系,四川 遂宁 629000;2.四川师范大学 化学与材料科学学院,四川 成都 610068)
共轭π电子体系的稠环芳烃是重要的一类有机半导体材料[1-2],其具有独特且可调控的分子结构、自组装行为和光电性质,在有机场效应晶体管,有机发光二极管,太阳能电池、化学传感器、锂离子电池固体电解质等领域中被广泛应用[3-6].尽管许多高性能p型有机半导体分子被合成,但是由于高的电子注入势垒能和有机阴离子在空气中的不稳定性,合成具有高电荷迁移率的n型有机半导体材料仍是当前电子学器件领域的难点[7].研究与开发有实用价值的有机半导体材料,提高其电学性能,特别是电荷迁移速率是关键[8-11].
六苯并蔻衍生物 (Hexa-peri-hexabenzo[a, d, g,j, m, p]coronene) 是一类具有较高本征迁移率的稠环芳烃材料之一,目前主要有两类异构体六苯并[bc,ef,hi,kl,no,qr]蔻[12-13]和六苯并[a,d,g,j,m,p]蔻[14-15].大共轭π电子体系的六苯并蔻衍生物实验合成较难,因此大部分六苯并蔻衍生物都是六烷基取代的衍生物.另外,它们较难与基底进行柱状单轴排列,通常利用脉冲辐射分解的时间分辨微波电导技术测试其电荷迁移速率.实验测试虽然测得了大量六烷基取代的六苯并蔻衍生物分子的电荷迁移速率[11,16],但是对分子结构与电荷迁移速率的变化规律认识尚浅.量子化学计算可以预测新型稠环芳烃分子的电荷迁移速率,从而进一步指导实验研究[17-21].例如,Kirkpatrick等[22-23]结合原子分子动力学模拟.Marcus-Hush理论和电荷动力学方程,将中间相结构的温度驱动变化与六烷基取代的六苯并蔻衍生物的柱状相中电荷迁移率的变化相关联,所有理论结果与时间分辨的微波电导测试数据相一致.宋岩等[24]利用密度泛函理论和Marcus-Hush电子转移理论探讨了多晶性对扭曲的六苯并蔻的电子结构和电荷输运性质的影响.我们也使用密度泛函理论研究不同拓扑结构的蔻类稠环芳烃分子的光电性能[25-27].
在分子层面,一般有机分子间排列紧密程度越高以及分子自组装有序度越高,其电荷迁移速率越高.因此,扩大芳烃共轭体系、芳环上引入杂原子、氟原子取代芳核上氢原子、引入不同取代基等分子设计手段,可显著改善相态稳定性和堆积有序度,提高电荷传输速率,改善材料在空气中的稳定性和器件的使用寿命,尤其在盘状分子硬核外围侧链中引入杂原子,可导致分子硬核中心由原来的富电性转为缺电性,如此分子构筑的盘状液晶在电荷传输上可能会由空穴传输转变电子传输,材料由p型向n型半导体转化.我们使用Gaussian 16 A.03[28]程序,利用密度泛函、杂化泛函理论和Marcus-Hush电子转移理论研究图1所示的母体六苯并蔻分子(A)和11个六苯并蔻衍生物分子(B~L)的电荷传输性能与取代基效应的关系,以期有助于实验合成电学性能优异的六苯并蔻衍生物.

图1 六苯并蔻及其衍生物分子结构式Fig.1 Molecular structures of hexabenzocoronene and its'derivatives
1 理论基础和计算方法
1.1 理论基础电荷迁移率可以由爱因斯坦方程计算得到:

式中,e和kB为常数,分别是电子电荷(1.60×10-19C)和波尔兹曼常数(1.38×10-23J/K),T为绝对温度,D是电荷以一个分子为起点向三维空间方向的平均扩散系数,可通过下式计算:

式中,ri和ki分别为相邻分子的距离和相邻分子间的电荷传输速率常数,pi为电荷向分子i迁移的几率.

共轭π电子体系的稠环芳烃分子具有一维电荷迁移的特性,其平均扩散系数可以简化为:

式中,r和k分别为相邻分子的盘间距和相邻分子间的电荷传输速率常数,将其带入(1)式可计算得到电荷迁移率:

根据Marcus电荷传输半经典模型[17,18,29-33],相邻分子间的电荷传输速率常数的表达式为:

式中,h和kB为常数,分别是Planck常数(6.626×10-34J·s)和Boltzmann常数,t为电荷传输矩阵元,T为绝对温度,λ为电荷传输重组能.一定温度下,影响电荷传输速率常数的主要参数为电荷传输矩阵元t和反应重组能λ.
重组能通过绝热势能面进行计算,即传输空穴的重组能λ+和传输电子的重组能λ-计算如下[17,18]:
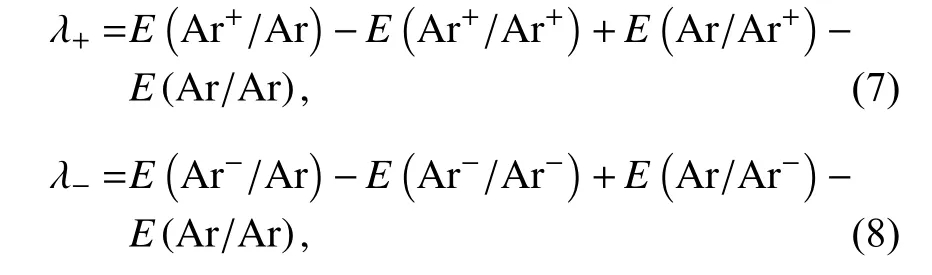
式中,“E(Ar+/Ar)”表示在Ar中性分子构型优化基础上的阳离子单点能,E(Ar+/Ar+)表示Ar+离子优化构型时的总能量,其余类似.
电荷传输矩阵元表征电子-电子相互作用的耦合强度,并且已经提出了几种方法评估分子二聚体内的转移积分.最简单的方法是前沿轨道能级分裂法[8,10],以质量中心为轴将分子和离子重叠,根据蔻类分子晶体衍射数据并结合支链与刚性核的二面角数据,将所研究12个分子的重叠距离定为0.35 nm[34].两个分子的相对扭转角θ范围定为0°~180°.通过在分子/分子阳离子系统中添加一个电子形成闭壳系统.计算HOMO和HOMO-1在过渡态的ΔHOMO能级分裂值,一半是空穴传输矩阵元t+.将分子/分子阴离子系统除去一个电子,则形成闭壳系统,然后计算LUMO和LUMO+1在过渡态的ΔLUMO能级分裂值,一半是负电荷转移矩阵元t-.空穴和电子的传递矩阵元可以通过将这些值减半来估计.在温度为298.15 K,根据Boltzmann分布,依据在不同二面角(Ei)和相应矩阵元 (ti),使用公式(9)计算不同旋转角度下12个分子离子重叠体系i的热力学概率ni.由公式(10)求得t2的热力学平均,
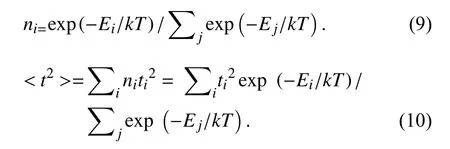
电荷转移反应的过渡态很难找到,因此几乎所有的研究工作都使用线性坐标来描述反应过程.反应中第i个内坐标Qi与反应坐标的关系为[36-37]:

Qip和Qir分别是平衡构型中产物和反应物的第i个内坐标.当R=0时,系统处于反应物平衡构型;当R=1时,系统处于产品的平衡构型;本文研究的反应是一种自交换反应,过渡态发生在R=0.5.
1.2 计算方法六苯并蔻及其衍生物分子的结构优化均采用Gaussian 16 A.03程序执行.为了确保计算的可靠性和优化构型的合理,分别采用目前电荷 传 输 性 能 研 究 相 对 准 确 的B3LYP[19,21-22,38]、M06-2X[21]和CAM-B3LYP[21-22]等3种密度泛函理论方法,且包含弥散函数和极化函数的基组6-311+G(d)进行结构优化,并在相同理论水平上进行频率分析,为确保势能面上所有的极小点没有虚频,SCF收敛标准采用10-8a.u.,结构优化中原子受力标准采用4.5×10-4a.u..计算HOMO与LUMO采用mulliken布局分析方法.
2 结果与讨论
2.1 分子结构优化使用Gaussian 16 A.03程序,在B3LYP/6-311+G(d)、M06-2X/6-311+G(d)、 CAMB3LYP/6-311+G(d)理论水平优化计算图1中A~L所示的—SH、—SCH3、—CH2SCH3、—CH3、—OH—OCH3、—NH2、—NHCH3、—N(CH3)2、—CN、—COOCH3等取代的12个六苯并蔻衍生物,得到极小点没有虚频的稳定结构.母体六苯并蔻(A)分子的结构优化图及部分原子的编号见支撑附件图S1.12个衍生物分子的部分二面角数据见附表S1.
由附表1的二面角数据可知,A分子中的蔻环平面性最好;其次是D、F、G和L分子,平面性相对较差的是B、C、H、I、J和K分子,这显然是源于取代基的影响.致使分子的刚性核蔻环存在不同程度的扭曲.图2给出了12个分子在CAM-B3LYP/6-311+G(d)理论水平下的计算优化结果.
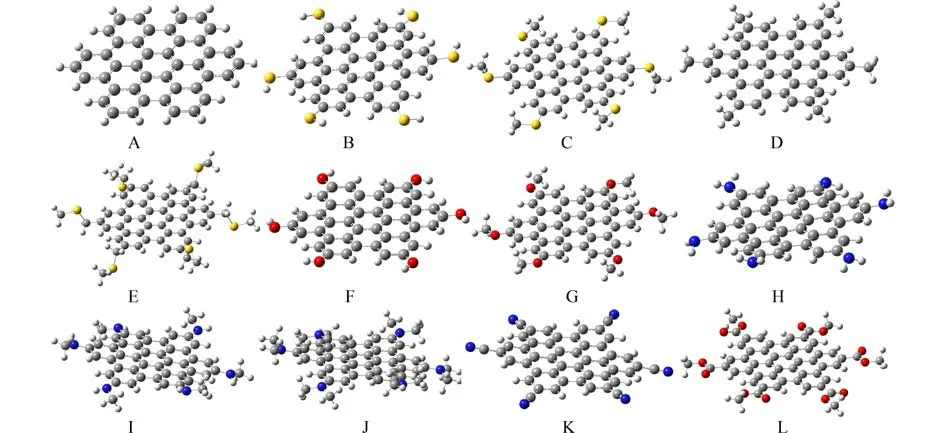
图2 六苯并蔻及其衍生物A~L的结构优化图Fig.2 Optimized structures of hexabenzocoronene and its' derivatives A-L
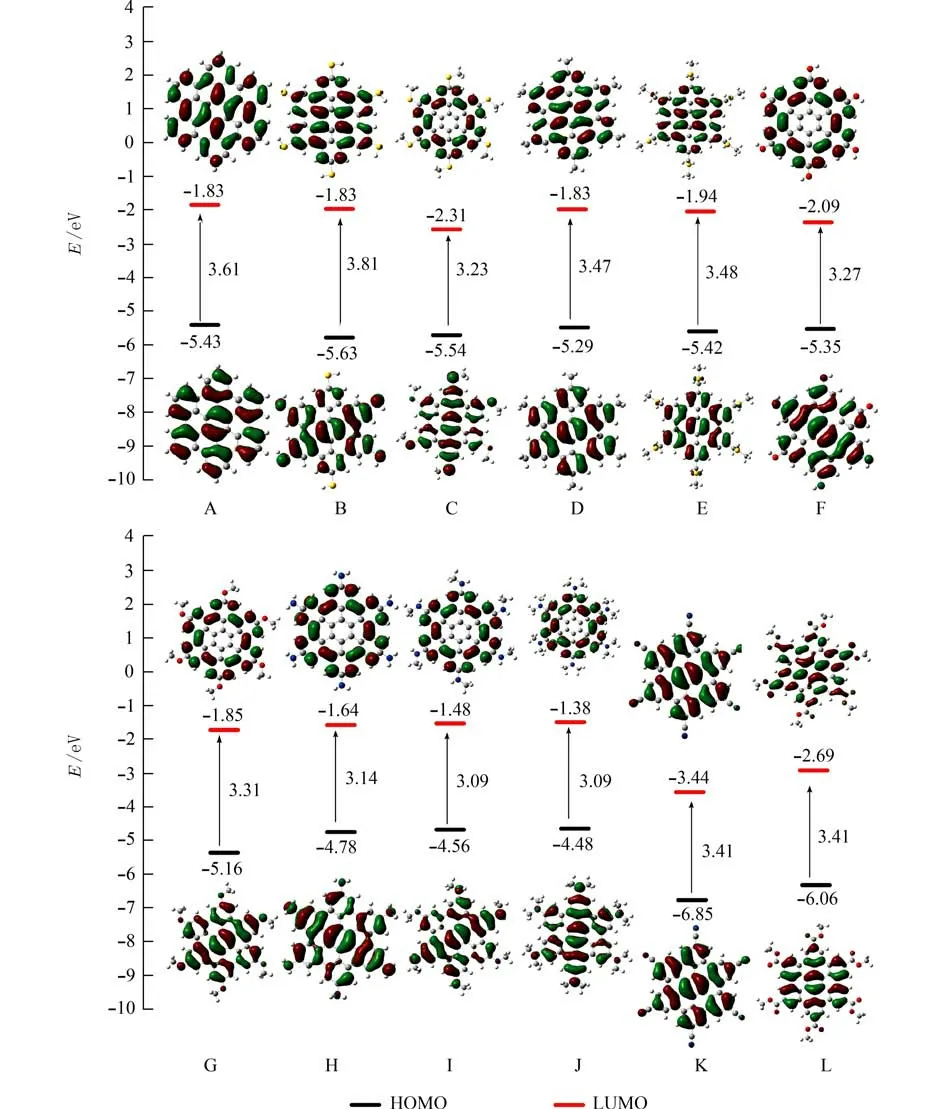
图3 六苯并蔻及其衍生物A~L的前线分子轨道图Fig.3 The frontier orbits of hexabenzocoronene and its' derivatives A-L
2.2 前线分子轨道和吸收光谱图3是在B3LYP/6-311+G(d)理论水平下计算得到12个分子的前线轨道本征能量、能隙及前线分子轨道图.由图3可知,12个蔻类分子的能隙在3.27~3.88 eV范围,位于有 机 半 导 体 的 能 隙 范 围1.4~4.2 eV (135.08~405.24 kJ/mol)[39]内,表明所研究的12个蔻类衍生物分子归属于有机半导体分子.与母体分子A相比,K、L分子的最高占据轨道 (HOMO)与最低空轨道(LUMO)的本征能量显著降低;B、C分子的最高占据轨道,C、E及F等3个分子的最低空轨道本征能量均明显降低;D、F、G、H及I等5个分子,其最高占据轨道本征能量均略有升高.除B分子外,相互协同作用使得分子的能隙均不同程度地变窄.尤其是C、F、H、I及J等5个分子的能隙值明显变小.原因可能是,在六苯并蔻环上引入S、O、N等杂原子后,分子的平面结构发生改变的缘故.预示在刚性核上引入—SCH3、—OH、—NH2—NHCH3及—N(CH3)2后,有利于电荷输运.
分子轨道在电荷转移中扮演十分重要的角色[18].分析图3可知, 12个蔻类衍生物分子,其HOMO电子云均是在六苯并蔻刚性核上均匀分布,与芳环相连的部分杂原子(S、N、O)的孤对电子对HOMO前线分子轨道有贡献;B、C、F、G、H、I及J等7个分子,其LUMO电子云密度分布相似,均定域在六苯并蔻刚性核的外环,呈现圆环形均匀分布; A、D、E、K及L分子,其LUMO与HOMO电子云相似,均在六苯并蔻刚性核上均匀分布.所有杂原子的孤对电子对LUMO前线分子轨道都没有贡献;由HOMO到LUMO的电子跃迁为π→π*和n→π*.
表1为在TD-B3LYP/6-311+G(d)理论水平下,计算所得到的吸收光谱数据.12个蔻类衍生物分子,其最低能量吸收峰波长在358~412 nm范围;在六苯并蔻上引入含杂原子(S、N、O)的取代基比引入含碳原子取代基,最低能量吸收峰红移更显著.主要源于这些杂原子的电负性远大于C原子,取代基对分子的吸引电子能力增强.所研究分子的最低能量吸收峰均源于S0→S5的HOMO→LUMO、HOMO→LUMO+1、HOMO-1→LUMO 及HOMO-1→LUMO+1的混合跃迁.
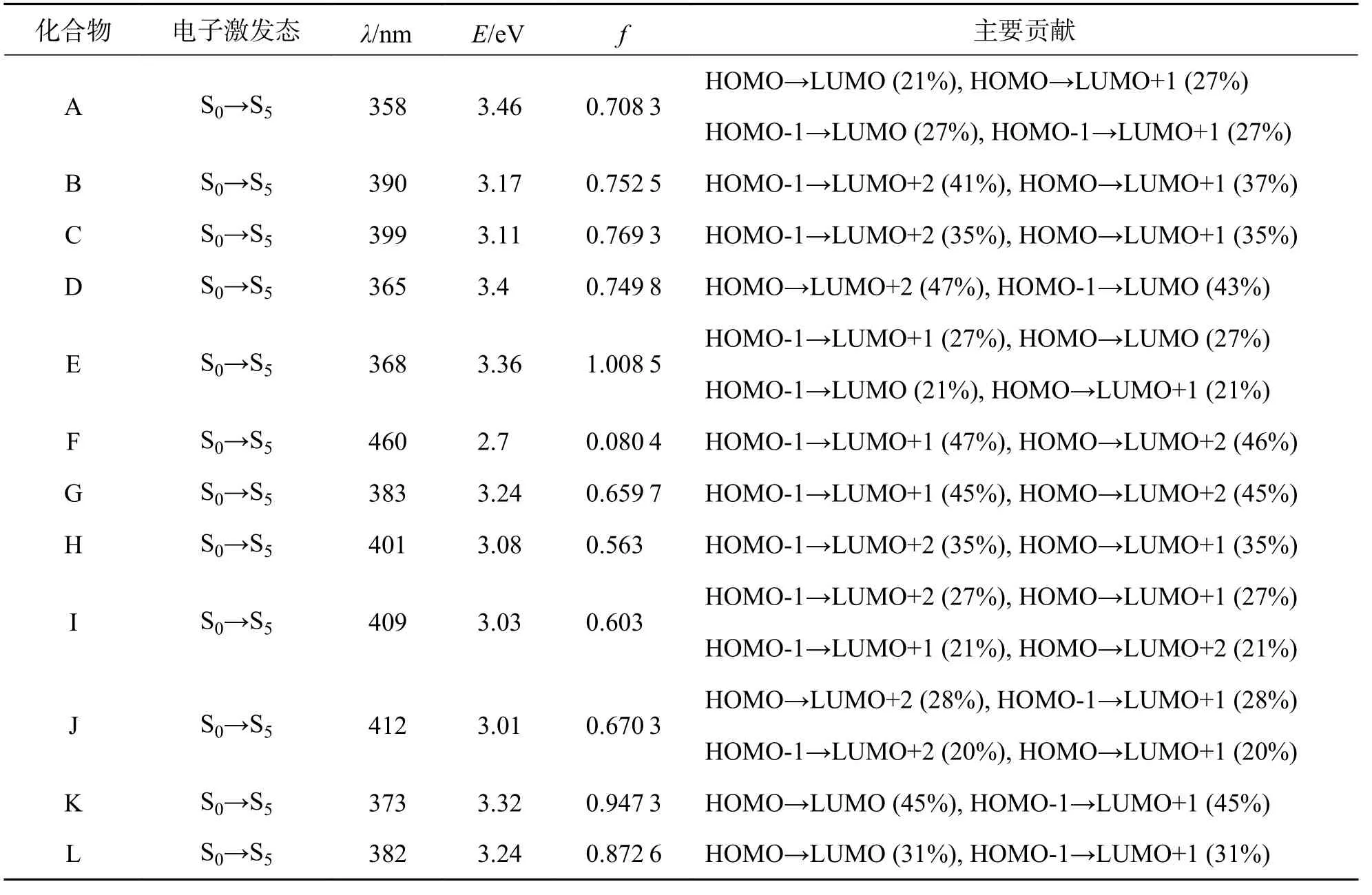
表1 在TD-B3LYP/6-311+G(d)理论水平下,计算所得到的吸收光谱数据Tab.1 The data of electronic absorption spectrum at the theoretical level of TD-B3LYP/6-311+G(d)
2.3 分子重组能采用6-311+G(d)基组,分别应用B3LYP、M06-2X及CAM-B3LYP密度泛函方法,优化得到的分子和离子能量、分子构型下的离子能量、离子构型下的分子能量列于支撑附件表S2,采用公式(7)与(8)计算得到12个带不同取代基的六苯并蔻分子的空穴(λ+)和电子传输重组能(λ-)列于支撑附件表S3,变化趋势见图4.
采用M06-2X与CAM-B3LYP方法计算得到的空穴和电子传输重组能接近,但采用B3LYP方法计算电子传输重组能时,对于B、C、K、L分子的计算结果,与前两种方法的计算值相比偏差较大.所研究的每个分子,由于13个相互内嵌的苯环盘状刚性核,所形成的柱状结构重叠面积较大.致使分子的重组能均比较小,更有利于电荷沿轴向传输.
由Marcus理论可知,电荷传输速率与重组能成反比.与母体A分子相比,E、F、G、H、I、J及L分子的空穴传输重组能增大,预示这些分子不利于空穴传输;B、C、F、G、H及I 分子的电子传输重组能略有降低,均小于或接近典型电子传输材料8-羟基喹啉铝(Alq3)的电子传输重组能 (0.276 eV)[40],预示这些分子有利于电子传输.

图4 六苯并蔻及其衍生物A~L的空穴与电子传输重组能Fig.4 Organization energies λ of hexabenzocoronene and its' derivatives A-L
2.4 空 穴 与 电 子 传 输 矩 阵 元在B3LYP/6-311+G(d)、M06-2X/6-311+G(d)、 CAM-B3LYP/6-311+G(d)理论水平下,计算得到的电荷传输矩阵元t数据见支撑附件表S4,变化趋势见图5.由图5可知,对于空穴电荷传输矩阵元(t+),3种方法计算结果均较为接近.CAM-B3LYP方法计算值略有偏大,但变化趋势相同,A、D及K分子的空穴电荷传输矩阵元(t+)明显较大,所有分子中D分子最大.对于电子传输矩阵元(t-),B3LYP与CAM-B3LYP两种方法计算结果均较为接近,M06-2X方法计算结果偏大,而变化趋势相同,与母体A分子相比,C、D、F、G、H、I、J及K分子的电子传输矩阵元增大,尤其是引入吸电子能力较大的N与O杂原子,所得到的G、H、I及J等4个分子增加幅度更大.由Marcus理论可知,电荷传输速率与传输矩阵元成正比,因而在六苯并蔻环上引入含N与O杂原子的取代基,有利于电子传输.
2.5 电 荷 传 输 速 率 常 数 和 迁 移 速 率由 公 式(1)和公式(6)分别计算12个分子的电荷传输速率常数k和迁移速率μ见表2,变化趋势见支撑附件图2.M06-2X方法计算值与B3LYP及CAM-B3LYP二种方法计算值偏差较大,但变化趋势相同.为此,我们以B3LYP与CAM-B3LYP二种方法的计算结果来进行讨论.
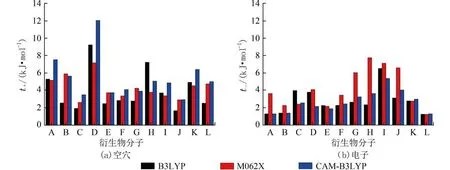
图5 六苯并蔻及其衍生物A~L的空穴与电子传输矩阵元Fig.5 Hole and electron transport matrix elements of hexabenzocoronene and its' derivatives A-L
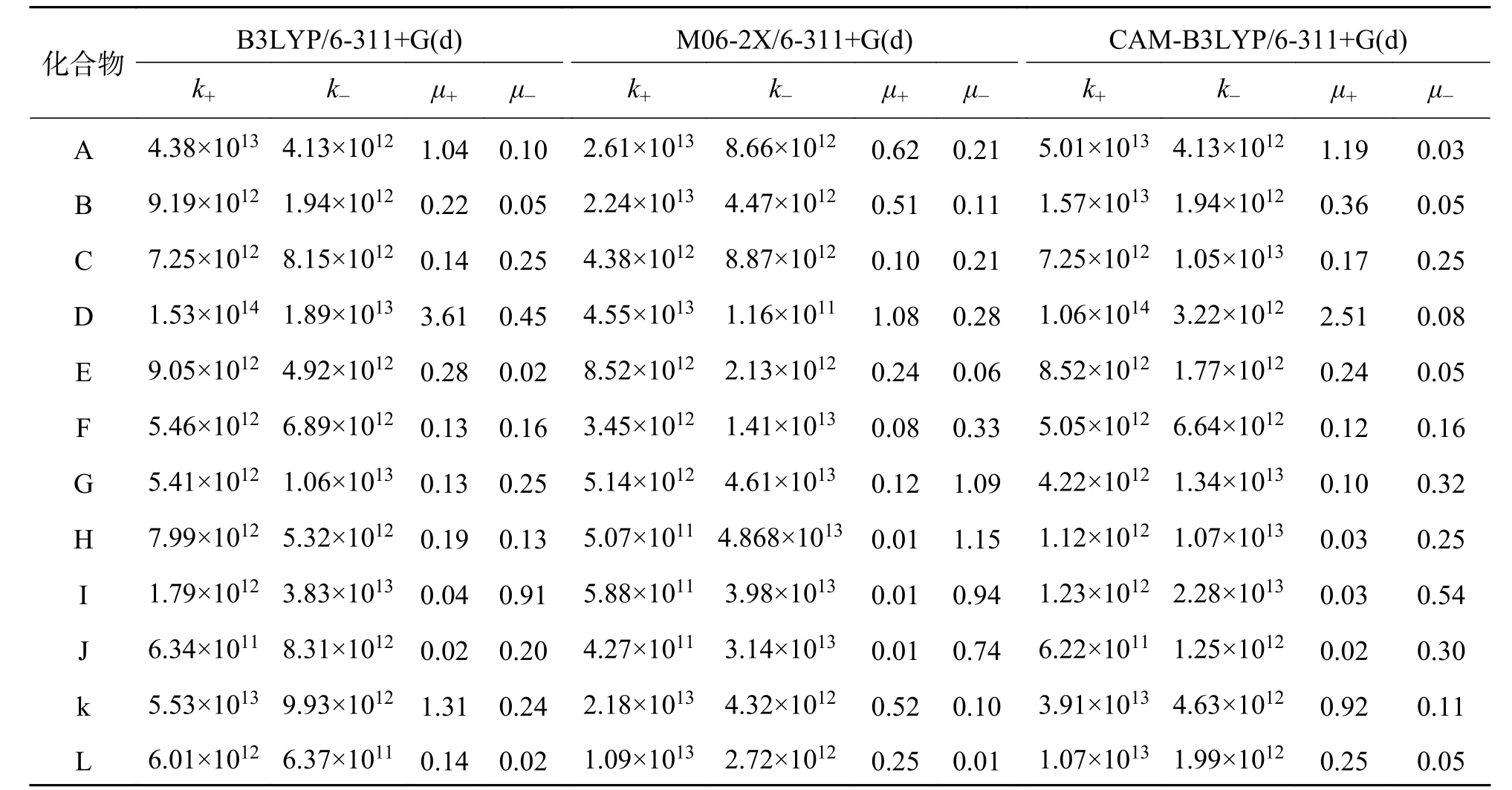
表2 六苯并蔻及其衍生物A~L的空穴与电子传输速率常数k和迁移速率μTab.2 Charge transfer rate constant (s-1) and transport rate μ (cm2·V-1·s-1) of hexabenzocoronene and its' derivatives A-L
母体A(六苯并蔻)、D、E及K分子的空穴迁移速率明显大于电子迁移速率,尤其是D、K 2个分子,D分子的空穴迁移速率是电子迁移速率的31倍,K分子的空穴迁移速率是电子迁移速率的8倍,这主要源于它们具有较小的空穴传输重组能和较大的空穴传输矩阵元.结果显示在六苯并蔻环上引入含碳原子的取代基后,不会改变半导体材料的电荷传输类型,有望设计为性能优良的p型有机半导体材料.与母体A分子相比,在六苯并蔻环上引入含电负性较大的杂原子取代基,所得到的C、G、H、I及J等5个分子,尽管电子传输重组能减小幅度不大,但电子传输矩阵元增大了2~4倍,二者协同作用使其电子迁移速率为空穴移速率的1.7~18倍.从而改变了半导体材料的电荷传输类型,由空穴传输为主(p型)转变为电子传输为主(n型)有机半导体材料,有望设计成性能优良的n型有机半导体材料.
3 结论
在 B3LYP/6-311+G(d)、M06-2X/6-311+G(d)、CAM-B3LYP/6-311+G(d)理论水平,分别对12个不同取代基的六苯并蔻分子的结构优化和电荷传输速率的量子化学计算.得到以下结论:
(1) 采用B3LYP与CAM-B3LYP泛函的计算结果接近,长程较正泛函CAM-B3LYP更适合研究目标体系的电荷传输性能.
(2) 在六苯并蔻环上,引入6个—CH3与—CN取代基,得到的D、K分子,空穴迁移率相对较大,分别为2.51 cm2·V-1·s-1和0.92 cm2·V-1·s-1,可设计为性能优良的p型有机半导体分子;引入6个—SH、—CH2SCH3及—COOCH3取代基,得到的B、E、L等3个分子,与母体六苯并蔻A分子相比,对分子的电荷传输速率改善较小;引入6个—SCH3、—OCH3、—OH、 —NHCH3、—N(CH3)2等取代基,所得到的C、G、H、I及J等5个分子,其电子迁移速率为空穴迁移速率的1.7~18倍,它们有望设计成性能优良的n型有机半导体材料.
猜你喜欢
车用发动机(2021年5期)2021-10-31
电源技术(2021年7期)2021-07-29
中国计算机报(2020年15期)2020-05-13
世界农药(2019年3期)2019-09-10
赤峰学院学报·自然科学版(2018年7期)2018-08-11
黑龙江电力(2017年1期)2017-05-17
中学生数理化·高二版(2016年3期)2016-12-26
CHIP新电脑(2016年9期)2016-09-21
合成化学(2015年10期)2016-01-17
华东师范大学学报(自然科学版)(2014年1期)2014-04-16
