
辽宁省首株柯萨奇病毒B组5型全基因组序列分析
2023-02-02 13:19孙英伟毛玲玲
微生物学杂志 2023年6期
雷 露, 于 伟, 张 倩, 王 博, 孙英伟, 毛玲玲
(辽宁省疾病预防控制中心,辽宁 沈阳 110005)
引起手足口病的肠道病毒主要为新型肠道病毒(Enterovirous)71型、柯萨奇病毒(Coxsackievirus)A组和B组、埃可病毒(ECHO)等,早期对手足口的病原学检测以EV71和CVA16为主,随着对手足口病病原的深入了解及检测技术的发展,人们把目光转向了非EV71、非CVA16肠道病毒基因型的研究。柯萨奇病毒B组5型(CV-B5)是柯萨奇病毒B组的一员,原型株为Faulkner,于1952年在瑞典首次分离[1]。由其引起的手足口病、心肌炎、病毒性脑炎、急性弛缓性麻痹等多种疾病在国内外均有流行:1999年至2011年韩国第三大常见肠道病毒[2];2000年至2004年法国第四高发肠道病毒[3];2009年山东省临沂市手足口病暴发疫情[4];2013年至2014年江苏省邳州市疱疹性咽颊炎病例中检测到CV-B5[5];2018年至2019年云南省昆明市手足口病和疱疹性咽颊炎病例中也检测到[6]。辽宁省历年监测数据显示[7-10],非EV71、非CVA16肠道病毒在我省手足口病发病病原谱中所占比例逐年增加,2015年首次超过50%,达到62.04%;2017年占比51.05%;2018年高达86.80%。这与北京[11]、安徽[12]、深圳[13]、武汉[14]等地手足口病的病原变化特征相似。鉴于国内外流行趋势,应加强对非EV71、非CVA16肠道病毒的监测,掌握辽宁省手足口病病原的构成变化及其他肠道病毒病原的遗传进化情况。CV-B5血清型的流行病学数据在全世界有限,国内还没有被纳入疾病监测系统,很难评估疾病负担。肠道病毒普遍存在重组现象,近年来引起手足口病的病原体也在不断变化,因此本研究对辽宁省首株CV-B5 分离株进行了全基因组测序,并对测序结果进行进化与重组分析,从分子水平解释病毒遗传变异及流行规律,希望能对辽宁省CV-B5 的分子流行病学研究和相关疾病的防控提供参考。
1 材料与方法
1.1 材料
1.1.1 标本 2018年辽宁省14个地级市疾控中心报告手足口病例的咽拭子及粪便标本。各市完成肠道病毒通用核酸检测后,阳性标本(粪便1 880份,咽拭子1 916份)一个月内送至辽宁省疾控手足口设备实验室。
1.1.2 主要试剂与仪器设备 人横纹肌瘤细胞(RD)为本单位冻存;DMEM、小牛血清为美国Gibco公司生产;Pen-Strep solution(青霉素、链霉素)为Biological Industries生产;提取试剂为硕世生物病毒核酸提取试剂盒(磁珠法)提取试剂盒。全自动核酸提取仪(SSNP-9600A,江苏硕世生物科技公司);PCR扩增仪(EDC-810,北京东胜);荧光定量扩增仪(Viia7,美国ABI);水平电泳仪(Bio-rad Mini-Sub Cell 1704469,美国伯乐);凝胶成像仪(Bio-rad GelDoc XR+,美国伯乐);使用HiScript II One Step RT-PCR Kit(诺唯赞)对CV-B5进行全基因组富集扩增。
1.2 方法
1.2.1 病毒分离 粪便和咽拭子核酸阳性标本经处理后接种RD细胞,在36 ℃和5%的CO2条件下培养5~7 d,每天在显微镜下观察,当75%的培养物出现细胞病变效应(Cytopathic effect,CPE)时,收获病毒培养物上清液进行肠道病毒分型的核酸检测,若连续两代均没有发生病变,则判断为阴性。样品处理和病毒分离培养具体操作参照《手足口实验室手册(2010第4版)》。
1.2.2 核酸提取肠道病毒检测与分型 病毒RNA使用硕世生物病毒核酸提取试剂盒(磁珠法)进行核酸提取。肠道病毒所有型别鉴定使用硕世生物荧光定量试剂盒进行核酸检测。
1.2.3 CV-B5全基因组RT-PCR扩增 使用HiScript II One Step RT-PCR Kit对CV-B5的全基因组进行富集扩增。参照文献[15]设计引物序列上游引物:TTAAAACAGCCTGTGGGTTGT;下游引物:ATTCTCCGCATTCGGTGCGG。反应体系:RNase Free Water 13 μL,5*QIAGEN One Step RT-PCR Buffer 5 μL,QIAGEN One Step RT-PCR Enyzme Mix 1 μL,Ribonuclease inhibitor(50 U/μL) 0.1 μL,上下游引物(20 μmmol/L)各0.5 μL,模板5 μL。反应条件:50 ℃ 30 min;95 ℃ 15 min; 95 ℃ 3 min,94 ℃ 30 s, 58 ℃ 30 s, 72 ℃ 1 min,40个循环;72 ℃ 10 min。凝胶电泳检测扩增产物。
1.2.4 序列测定和分析 用Qubit进行定量分析后送至上海伯杰医疗科技有限公司进行高通量测序,测定后返还拼接序列。拼接序列通过NCBI网站进行在线BLAST比对,在GenBank上下载CV-B5流行株、原型株和主要血清型的全基因组序列(参考株GenBank登录号见图1)进行同源性分析和进化分析,基因进化树利用MEGA 5.2软件构建,采用邻接法,自展值设置为1 000。

图1 柯萨奇病毒B组5型分离株全基因组序列系统进化树Fig.1 Phylogenetic tree based on complete genome sequences among Lianing and other coxsackievirus B5 strains
1.2.5 重组分析 重组分析采用RDP4和SimPlot 3.5.1软件,选择默认参数。
2 结果与分析
2.1 全基因组测序及核酸序列同源性分析
将分离到的CV-B5毒株(LN2018-23-21/CHN/2018)进行全基因序列扩增,测序拼接后得到的CV-B5基因组全长7 357 bp,GC含量47.66%,包括5′非编码区(5′-untranslated region,5′-UTR),3′-UTR和一个编码2 307个氨基酸的开放读码框。5′端非编码区(5′-UTR)长720 bp,与病毒的复制和翻译有关;3′端非编码区(3′-UTR)长100 bp,与病毒RNA的合成有关。编码区为6 558 bp,由P1、P2、P3三个部分组成,P1区负责编码结构蛋白,P2、P3区负责编码非结构蛋白。P1区可分为 VP4、VP2、VP3 和 VP1,VP1区蛋白包含多个抗原结合位点,是肠道病毒分子分型的主要标志。P2 区 分 为 2A、2B 和 2C,P3 区 分 为 3A、3B、3C 和3D,它们负责编码各种酶和功能蛋白。测序结果在美国生物技术信息中心网站进行BLAST分析,显示序列与CV-B5同源性最高。
2.2 预测蛋白序列及同源性分析
将辽宁分离株与原型株、国内流行株进行核苷酸和氨基酸同源性分析。该流行株与国内流行株的全基因组核苷酸序列同源性为78.5%~98.8%,氨基酸序列同源性为75.3%~96.7%,与山东分离株MN541028/SD/CHN/2018同源性最高,与原型株全基因组核苷酸序列同源性为78.9%,氨基酸序列同源性为76%,与CV-B5原型株VP1区序列对比发现,该分离株在第7、19、91、125、132、156、180、268、273、276和279共11处位点发生了替换(表1)。

表1 辽宁柯萨奇病毒B组5型(CV-B5)分离株VP1变异位点Table 1 VP1 mutation of coxsackievirus B5 (CV-B5)strains isolated in Liaoning
2.3 进化分析
通过MEGA5.2软件(neighbor-joining)对辽宁分离株和不同国家、地区在不同年份的流行株进行系统进化分析。从GenBank上下载36株CV-B5基因组全长序列作为参考株,将LN2018-23-21/CHN/2018与参考株序列基于全基因组核苷酸序列构建种系进化树(图1)。
进化树分析结果表明,可将CV-B5流行株划分为A~D四个基因型。基因型的划分体现了CV-B5在地理和时间上的分布差异。A基因型只包含原型株Faulkner;B基因型包含2000年韩国分离株;C基因型则是中国大陆2008年至2015年的主要优势流行基因型,此外还包括两株日本流行株(2011年、2015年)和1株2015年美国流行株;D基因型主要为国外流行株及国内2015年以后的流行株,包括2株2015年瑞士分离株,3株2013年至2017年美国分离株,3株2008年至2019年日本分离株,3株2008年至2017年澳大利亚分离株,3株2018年山东分离株及本次辽宁分离株。
2.4 重组分析
为了确定LN2018-23-21/CHN/2018在其进化过程中是否与其他肠道病毒B群发生重组,在GenBank上选取了同源性最高的基因序列,通过RDP软件,运用4MaxChi、RDP、BootscanChimaera、SiScan和3Seq方法分析,发现LN2018-23-21/CHN/2018有三处重组。第一处开始断裂点在697,结束断裂点为832(MaxChi法,P=2.148×10-2),亲本序列为KP289438/BEIJING/CHN/2013和AF114383/Faulkner;第二处开始断裂点在1 164,结束断裂点为3 326,(RDP法,P=1.400×10-15),亲本序列为M16572/CV-B3和JX017382/Henan/CHN/2011;第三处开始断裂点在5 916,结束断裂点为7 380(3Seq法,P=1.831×10-14),亲本序列为AF114383/Faulkner和AF081485/CV-B2。
为进一步验证重组分析结果的可靠性,运用SimPlot 3.5.1软件以LN2018-23-21/CHN/2018为质询序列,对辽宁分离株、亲本序列及其他EV-B原型株序列进行相似性分析(图2)。结果显示,在P3区的3D区段,与AY875692显示出约70%以上的相似性,在6 400左右相似度迅速下降,最低时仅为50%。
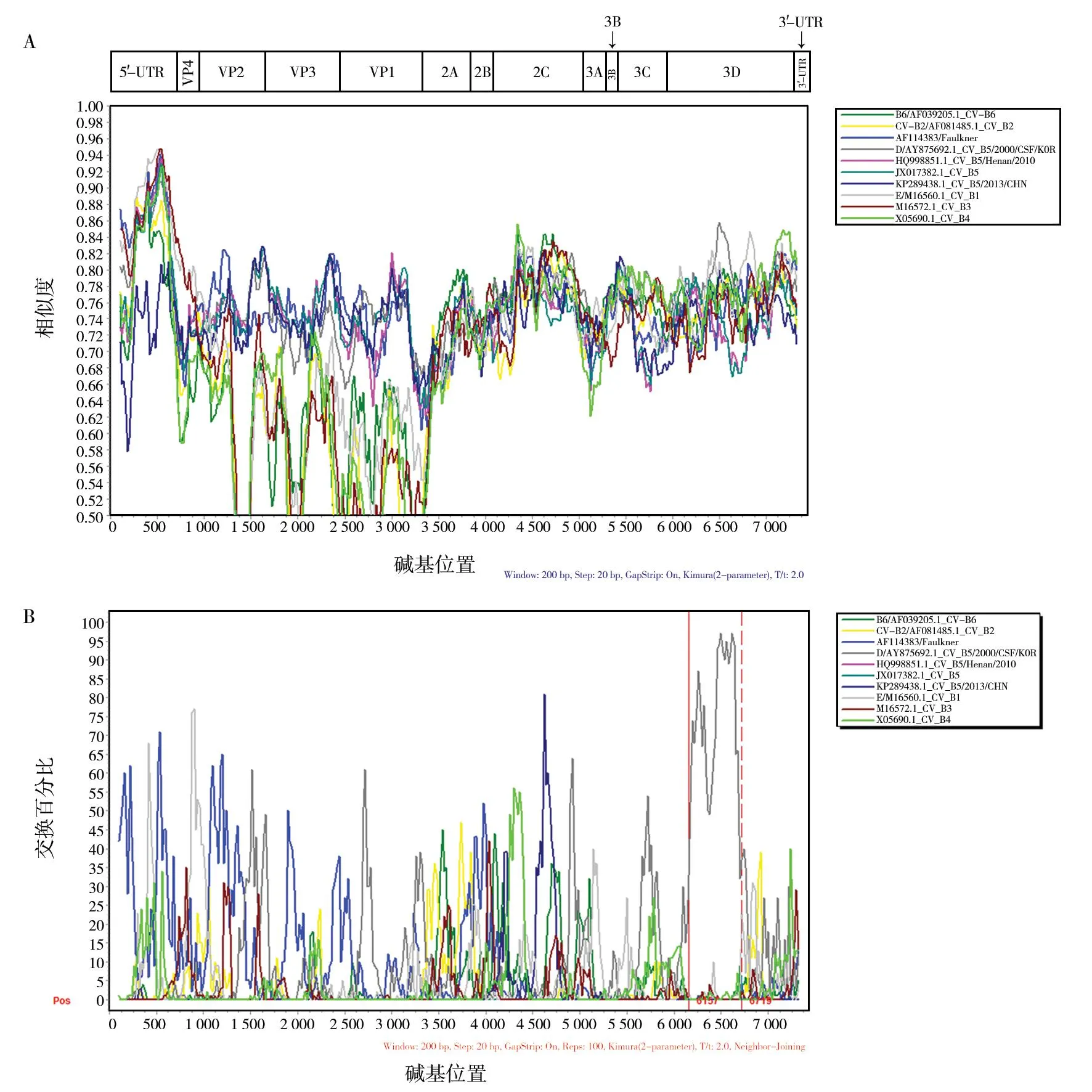
图2 辽宁分离株(LN2018-23-21/CHN/2018)与柯萨奇病毒B组肠道病毒主要血清型原型株序列重组分析Fig.2 Recombination analysis based on complete genome of LN2018-23-21/CHN/2018 and enterovirus B strainsA:LN2018-23-21/CHN/2018 Similarity plot分析;B:LN2018-23-21/CHN/2018 bootscanning分析A:LN2018-23-21/CHN/2018 Similarity plot analysis;B:LN2018-23-21/CHN/2018 bootscanning analysis
3 讨 论
CV-B5作为柯萨奇病毒B组的成员,属单股正链RNA病毒,RNA病毒会因为缺乏聚合酶修正功能而易引起基因突变,具有高进化率特点[16]。在编码结构蛋白的P1区域,尤其是编码主要中和抗原位点的VP1区,氨基酸序列较为保守,进化方式主要是点突变[17]。从进化分析结果看,与CV-B5原型株Faulkner的全基因组核苷酸序列同源性为78.9%,氨基酸序列同源性为76%;与CV-B5原型株Faulkner的VP1氨基酸序列相比,辽宁分离株共有11处发生替换。与VP1区相比,全基因组的同源性相对较低,这可能与肠道病毒的重组事件通常发生在非结构性编码区有关。与原型株相比,辽宁分离株在VP1区发生的替换中,7、91、125、132、268和273这六处和天津市2014年至2016年流行株的替换位置和替换序列一致[18]。VP1区因其与抗原性密切相关,这段区域发生氨基酸替换极有可能影响蛋白质空间构象,导致毒力变化[19],这也是造成病毒暴发和流行的主要原因。据报道,19、156、180、276四处氨基酸变异位点还是区分基因型C型和D型的特征性位点[20],本次分离株在这四个位点均发生了变异,这与本研究基因分型结果相互吻合、相互印证。
依据肠道病毒和CV-B5的分型标准,本研究将CV-B5流行株划分为A~D四个基因型。C 基因型是我国主要优势基因型[19],以中国大陆2008年至2015年的主要优势流行基因型为主,此外还包括两株日本流行株(2011年、2015年)和一株2015年美国流行株。D基因型国内以2015年以后的流行株为主,本次辽宁分离株属于D基因型,与2018年山东一分离株同源性最高,推测是由于人口流动造成的毒株跨地区传播事件。而与我国其他地区分离的CV-B5毒株核苷酸和氨基酸序列存在一定差异,提示随着时间的推移,CV-B5毒株已经发生一定程度的变异。
病毒进化有多种进化方式,基因重组是一个重要途径,基因重组所形成的病毒的遗传变异,远比单纯由突变造成的变异所引起的变化更大[21]。基因重组事件频繁发生于肠道病毒的非结构蛋白编码区,如云南分离株 A210在 3D区与CV-B3发生重组,河南分离株19CSF与吉林分离株CC10在2C区与CV-B3发生重组,河南分离株在P3区与CV-B4发生重组[21-25]。本研究通过RDP4重组分析,获得可能产生的重组信息,再采用SimPlot3.5.1软件进行相似性分析,对潜在重组信号的可靠性检验加以验证。结果表明,辽宁分离株在6 157~6 719 bp位点与AY875692 CV-B5/CSF/KOR/2000发生重组。基于CV-B5基因组结构分析,此段重组位于P3的3D区,属于非结构编码区,与以往报道的引发HFMD的CV-B5毒株重组位置一致[25-26]。结合国内关于CV-B5重组的研究[27-30]可以看出,重组事件的发生在国内CV-B5分离株中是一种常见现象。病毒的进化会导致毒力改变、传播力增强、组织亲嗜性改变,重组毒株可能会使原来一些非致病或低致病病原体变为高致病性病原体,造成疾病的传播,增加疾病负担。
辽宁省历年手足口病病原监测情况结果表明,非EV-71、非CA-16的其他肠道病毒的检出持续上升。加大CV-B5的监测力度,开展全基因组测序和分析,有助于了解肠道病毒的基因特征和进化规律,为手足口相关疾病的防控提供参考。
猜你喜欢
中国生殖健康(2020年7期)2021-01-18
幼儿园(2020年18期)2020-12-30
家庭医学(下半月)(2020年7期)2020-08-24
中国医药指南(2017年3期)2017-11-13
安徽医科大学学报(2016年12期)2017-01-15
现代检验医学杂志(2015年6期)2015-02-06
现代检验医学杂志(2015年6期)2015-02-06
实验动物与比较医学(2014年5期)2014-02-28
中医研究(2013年5期)2013-03-11
中国糖料(2013年1期)2013-01-22
