
负载贵金属的γ-Al2O3 催化剂的抗烧结策略
2023-01-21 00:57:08彭胜攀马子然马静王红妍敖志敏李永龙王宝冬
工程科学学报 2023年2期
彭胜攀,马子然,马静,王红妍,敖志敏,李永龙✉,王宝冬✉
1) 北京低碳清洁能源研究院,北京 102211 2) 广东工业大学环境科学与工程学院,广州 510006
煤化工行业尾气排放产生的挥发性有机物(VOCs)种类繁多、浓度低、风量大[1–2],催化燃烧技术可将该工况下VOCs 氧化成无害的CO2和H2O 并实现达标排放.VOCs 氧化催化剂设计与制备是催化燃烧技术的关键[3].VOCs 氧化催化剂一般由载体和贵金属(活性组分)两部分组成.根据“催化燃烧法工业有机废气治理工程技术规范(HJ 2027—2013)”,催化剂应能够耐受900 ℃短时间高温冲击以及8500 h 以上的稳定运行.当VOCs氧化催化剂负载同样种类和质量的活性组分后,催化活性取决于贵金属分散度和贵金属与载体间的相互作用.相比其他材料作为载体,(1)γ-Al2O3的较高比表面积(100~300 m2·g–1)为负载的贵金属提供了较大的分散面积,有利于提高贵金属分散度;(2)将γ-Al2O3置于700~800 ℃下一定时间,仍可保持晶相稳定,确保载体蓬松多孔,避免了活性组分被包埋降低贵金属利用率;(3)γ-Al2O3界面上存在丰富的不饱和配位铝离子形成L 酸位点,起到锚定贵金属作用并调节贵金属d 轨道的电子特性,降低载体表面贵金属迁移和团聚从而提高贵金属位点的催化活性;因此工业上一般用γ-Al2O3作为VOCs 氧化催化剂载体[4].
一方面,当γ-Al2O3暴露在高于800 ℃环境中,其晶型会逐渐发生转变并随着温度升高至1200 ℃后形成α-Al2O3,晶相转变导致氧化铝载体比表面积下降和载体界面性质的变化(不饱和配位铝离子)[4];另一方面,负载的贵金属也会随着温度升高、运行时间增长、气氛变化(水汽等)等因素发生贵金属团聚和烧结[5].载体晶相变化和贵金属团聚不仅降低暴露的贵金属活性位点数量,还弱化了载体与贵金属间相互作用,降低单个活性位点的本征活性,从而加速了催化剂失活,降低催化剂使用寿命.本文对当前VOCs 氧化催化剂失活机制进行阐述,在此基础上对抑制或延缓负载于γ-Al2O3的贵金属及γ-Al2O3失活的方法进行梳理和总结,从稳定性的角度提供VOCs 氧化催化剂设计策略并对未来VOCs 氧化催化剂发展方向进行了展望.
1 催化剂失活机制
催化燃烧技术一般采用整体式催化剂,结构如图1 所示.利用惰性氧化物烧制出孔道平行且相互不连通的蜂窝陶瓷作为催化基底(如堇青石2MgO·Al2O3·5SiO2),将催化剂粉体涂覆在蜂窝陶瓷上形成几十到几百微米厚的多孔涂层.实际工况下,整体式催化剂体积空速在20000~25000 h–1范围内,且气氛湿度较高(煤化工脱碳再生单元尾气湿度在10%~19%),高湿度、高空速对整体式催化剂性能和寿命产生重要影响.
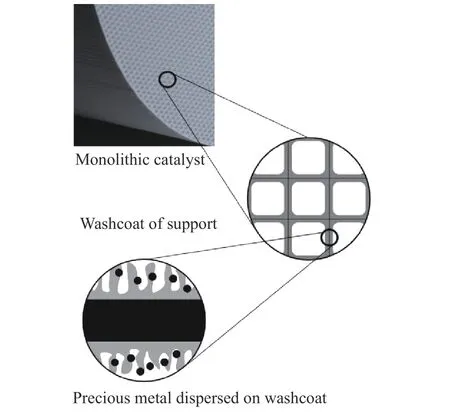
图1 整体式VOCs 氧化催化剂结构Fig.1 Structure of monolithic VOCs oxidation catalyst
催化剂失活有多种原因,根据催化剂结构变化可大致分成三种类型:贵金属团聚、载体烧结、贵金属中毒.如图2(a)所示,理想催化剂应保证在实际工况下长时间运行时,贵金属能够在载体表面均匀分散且不发生团聚,载体孔道保持通畅不发生阻塞,确保反应气体与贵金属充分接触.在催化剂使用过程中,载体和贵金属不可避免的发生烧结失活的现象(图2(b)和图2(c)),反应气氛中含有某些成分(如粉尘、硫化物、卤素等)也会导致贵金属中毒,如图2(d)所示,抑制甚至消除了贵金属的催化活性[6].
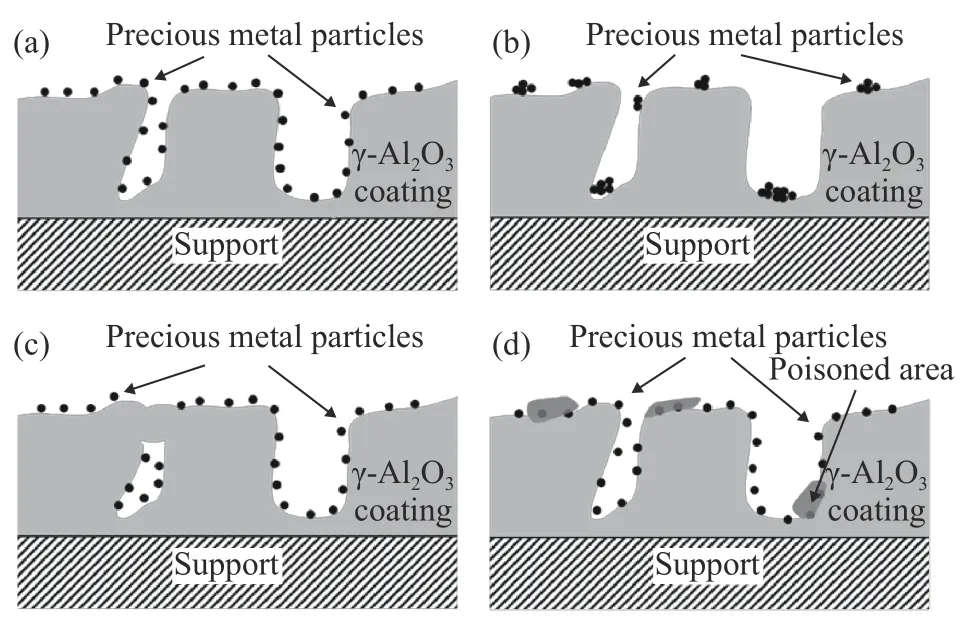
图2 整体式VOCs 氧化催化剂失活类型.(a)理想型;(b)贵金属烧结;(c)载体烧结;(d)中毒失活Fig.2 Types of deactivations of monolithic VOCs oxidation catalysts:(a) ideal catalyst;(b) sintering precious metal of catalysts;(c) sintering γ-Al2O3 of catalyst;(d) poisoned catalyst
1.1 贵金属烧结现象及成因
贵金属的用量、分散度对催化剂活性有决定性作用.高温导致分散于载体表面的贵金属颗粒逐渐变大,分散度下降,从而导致催化剂活性降低.贵金属烧结机理主要为:(1)Ostwald 熟化理论,均匀分散于载体上的贵金属颗粒大小并不相同,由于界面能作用使得较小颗粒消失而大颗粒逐渐增大;(2)颗粒迁移与团聚,当贵金属颗粒增长到一定尺寸并高于某一温度时,会以类似液态形式存在于载体上并发生布朗运动,不同尺寸的颗粒碰撞会使二者由于界面张力作用发生融合并进一步变大.
温度是贵金属烧结的主要因素之一.如表1所示,常用作催化活性组分的贵金属熔点温度(Melting point,Tm)都能够达到1000 ℃以上,但VOCs 催化转化温度最高一般不超过900 ℃却导致贵金属烧结.这是由于当贵金属达到许蒂希温度(Hüttig temperature,0.3Tm)时,贵金属原子在界面上振动加剧并加强了贵金属的扩散程度,以单原子、团簇、纳米颗粒形式存在于新鲜催化剂上,贵金属发生Ostwald 熟化并逐渐聚集形成较大颗粒.当温度升至塔曼温度(Tammann temperature,0.5Tm)时,贵金属微晶处于准液体状态并在载体表面发生明显迁移,与其他贵金属颗粒接触后融合,尺寸进一步增大[7].以Au 为例,当温度高于128 ℃时,已满足Au 颗粒团聚的热力学条件;VOCs 催化氧化温度一般在200~500 ℃之间,导致Au 容易团聚并失去活性,这是其成为应用于VOCs 催化氧化的难点之一.此外,相比块体或粉体材料,贵金属纳米颗粒或团簇具有更高的表面能及更低的熔点温度,随着热催化温度升高及使用时间延长更容易发生高温烧结现象.张静静等[8]曾报道,在室温或低温下储存的Au/CeO2有纳米金颗粒团聚的现象发生.
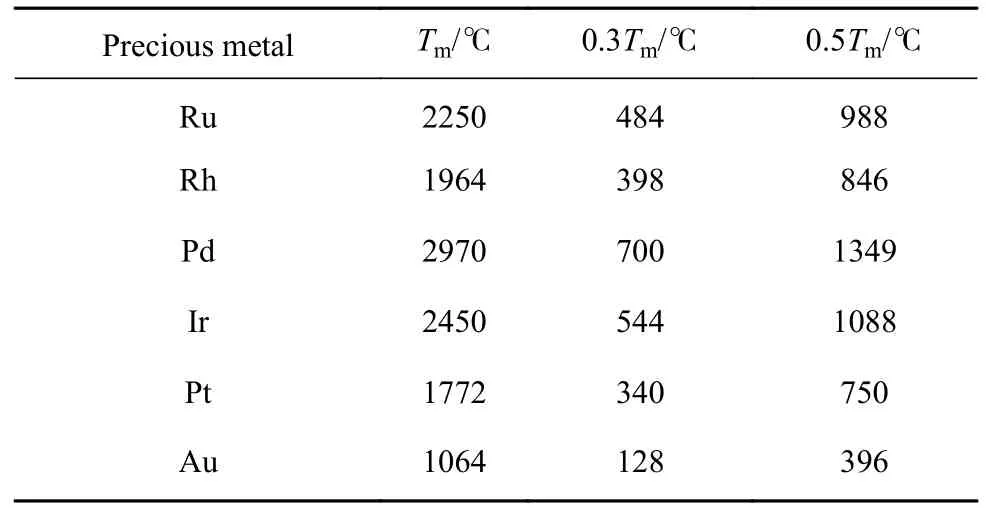
表1 贵金属单质特征温度Table 1 Characteristic temperature of precious metal
气氛对催化剂烧结也有一定影响.在催化氧化过程中,氧气在一定温度和浓度下会使得贵金属部分氧化降低晶体熔点,曾有文献报道[9]Pd 在还原性气氛中的烧结速率加快.在空气氛围下800 ℃老化Pt/Al2O3和Pt/MgAl2O4,载体上Pt 形成气相PtO2加剧了老化速度,老化5 h后,Pt 活性损失90%以上,30 h 后 Pt 团簇消失变为约200 nm 颗粒[10].蒋斌峰等[11]报道,含氯有机物在与贵金属催化剂反应过程中使活性组分从载体表面脱落和流失,减少了表面活性位点.
1.2 γ-Al2O3 烧结现象及成因
γ-Al2O3被作为催化剂或者催化剂载体而广泛应用,这是由于γ-Al2O3具有高的比表面积和较好的热稳定性,在700~800 ℃下仍能够保持晶型稳定.当温度高于800 ℃时,γ-Al2O3烧结使得比表面积下降、孔道变窄或堵塞导致分散的贵金属位点被载体包埋而失去了催化活性(如图2(c)所示).γ-Al2O3孔道变窄还会影响反应分子在孔道内的扩散和传质速度,随着在高温下使用时间延长,导致氧化铝晶相变化[12].整体式催化剂一般采用涂覆工艺将多晶的γ-Al2O3粉体粘附在堇青石等支撑体上,粉体中晶粒较小具有较高表面能[13]且堆积形成疏松多孔结构使得氧化铝在450 ℃就逐渐开始发生结构变化,并在750 ℃形成新的晶相[14].虽然γ-Al2O3具有一定的热稳定性,但长时间暴露在高温下,会逐渐发生晶相转变、烧结并导致催化剂不可逆失活.
1.3 中毒导致的烧结现象及成因
一般待处理气氛组成中含有的某些成分会导致催化剂中毒.如图2(d)所示,气氛中含有的粉尘、水汽等覆盖于活性位点上,使得催化剂可逆失活[15–16].当气氛中含有硫化物会导致载体富集硫酸盐改变载体活性[17],或者与贵金属形成硫化物、硫酸盐使得催化剂活性下降[18–19].此外碳、卤素、钒、钾及重金属元素等在催化剂表面富集也是催化剂失活的重要原因[20],尤其碱金属或碱土金属的引入会加速γ-Al2O3的烧结,降低比表面积[21].
2 催化剂抗烧结策略
针对催化剂在VOCs 降解反应中的热致失活现象,从四个方面梳理催化剂抗烧结的方法:(1)对γ-Al2O3载体表面改性改变晶相或引入其他元素提高载体熔点温度;(2)对贵金属颗粒表面修饰降低界面能;(3)空间限域避免发生团聚;(4)调控贵金属–载体相互作用.
2.1 γ-Al2O3 表面改性策略
γ-Al2O3具有高比表面积,表面有羟基基团、丰富的Lewis 酸和Bronsted 酸活性位点、五配位Al 物种,这些特点使其被广泛用作催化剂或者催化剂载体.γ-Al2O3作为催化剂或载体在高温下长期使用会发生相变致使比表面积骤降等,催化剂活性大幅降低[4].因而如何提高γ-Al2O3的热稳定性将是研究者持续关注和研究的焦点.
如Wu等[21]在制备γ-Al2O3前驱体时,将P 元素引入体系内在γ-Al2O3表面与羟基脱水缩合形成Al–O–P–O–Al 化学键,使得其热稳定性得到提升.当表面无P 元素,γ-Al2O3表面存在大量Al–OH 官能团,随着温度升高,相邻羟基之间发生脱水缩合形成Al–O–Al,导致界面应力随着温度升高发生表面折叠和聚集,最终导致载体烧结比表面积降低;而引入的PO43–具有稳定结构,与表面Al3+脱水形成Al–O–P–O–Al,增加相邻Al3+间键长,降低界面应力提高了γ-Al2O3界面热稳定性.但是,PO43–的引入降低了表面羟基的数量,相比未掺杂P 的γ-Al2O3,催化剂活性有一定程度降低.
在γ-Al2O3表面引入其他金属元素,在界面上形成高熔点晶型,亦可有效抑制体相发生变化导致的比表面积降低问题.Chen等[22]将Mg 引入γ-Al2O3形成高温稳定的MgAlO2尖晶石结构;引入稀土元素,如La[23],在表面形成LaAlO3钙钛矿结构,其晶体熔点达到2000 ℃以上,表面掺杂La 的钙钛矿结构使得贵金属能以单个离子状态存在于该晶相内形成稳定结构[24],提高VOCs 氧化催化活性.此外,α-Al2O3可耐受1000 ℃以上高温,随着高比表面α-Al2O3制备方法的开发[25–26],α-Al2O3亦可成为替代γ-Al2O3的选择之一.
2.2 贵金属修饰策略
2.2.1 壳层保护法
核壳结构纳米催化剂能够抑制催化剂活性组分团聚,确保高温环境下活性组分的分散[27–28].如SiO2具有化学性质稳定、熔点高等特点,将其作为活性组分壳层结构组成能保证催化剂的热稳定性和活性.如图3 所示,Habibi等[29]将7 nm 贵金属颗粒包覆于粒径为60 nm 的SiO2颗粒内,3.4 nm SiO2壳层确保了贵金属颗粒之间不接触,经过550 ℃和水汽的物质的量分数为5%老化处理后,仍可保持甲烷催化氧化效率不变.将包覆贵金属的二氧化硅颗粒锚定在γ-Al2O3上不仅提高了贵金属的水热稳定性,还对其催化效果起到促进作用.Zou等[30]将核壳结构PdO@SiO2负载于γ-Al2O3上在800 ℃仍可保持稳定;在380 ℃即达到90%甲烷转化率,相比PdO/γ-Al2O3活化能降低了19 kJ·mol–1,这是由于PdO@SiO2与γ-Al2O3载体界面间作用增强提高了贵金属分散性,SiO2包覆在PdO 活性中心提高了生成物(水分子)的脱附速率.Zhang等[31]采用一锅法制备负载Pd 的分子筛Pd@S-1,微孔结构限制了贵金属颗粒的长大以及移动,不仅具备高的活性和水热稳定性,还表现出优异的抗SO2中毒性能.
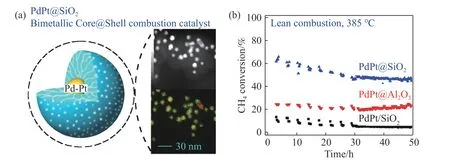
图3 贵金属颗粒包覆SiO2 示意图及性能对比.(a)Pt–Pd 合金被二氧化硅包覆形成核壳结构;(b) 核壳结构提高甲烷催化性能[29]Fig.3 Schematic showing the encapsulated PdPt@SiO2 catalyst and its hydrothermal stability for lean CH4 combustion: (a) Pt–Pd alloy is coated with silica to form a core-shell structure;(b) core-shell structure improves the catalytic performance of methane[29]
壳层作为贵金属颗粒保护层,既能够提高催化剂稳定性,又可调节贵金属与壳层间相互作用,进而改变贵金属活性中心活性.Ozawa等[32–33]将0.1% Pt 分别负载于CeO2和核壳结构的CeO2@ZrO2上,通过H2-TPR 发现CeO2@ZrO2的出峰数量和位置都与CeO2上Pt 物种差异明显,且当丙烷和丙烯转化率为80%时,Pt/CeO2@ZrO2比Pt/CeO2转化温度低了50 ℃左右.
2.2.2 贵金属熔点提升法
若提高贵金属相的熔点温度,其0.3Tm和0.5Tm相应增高,在同样温度环境下可提高贵金属的热稳定性,降低甚至避免贵金属颗粒的团聚.如表1所示,贵金属熔点高低不同,可与高熔点物种形成合金相,如Pd 比Pt 熔点温度高约1200 ℃,Pt–Pd合金比Pt 单质具有更高熔点温度,不仅提高了Pt 热稳定性,而且降低了Pt 氧分压抑制PtO2形成,降低了Pt 从催化剂上挥发的速度[34].Xiong等[35]发现,在Pt–Pd 合金中,当Pd 形成PdO 对降低Pt烧结速度起到了关键作用.Carrillo等[36]针对上述现象进行了机理探讨(如图4 所示),气相PtO2促进Pt–Pd 合金烧结过程.在800 ℃空气氛围老化Pt–Pd 合金,PtO2从合金相中挥发形成类似Pt 在外层的核壳结构及PdO 颗粒,PdO 捕获PtO2并固定在PdO 颗粒上.最终,在氧化气氛下Pt 逐渐从合金相中逃逸,Pd 单质形成PdO 颗粒成为锚定Pt 的场所.当经过还原处理后,再次形成Pt–Pd 合金颗粒.
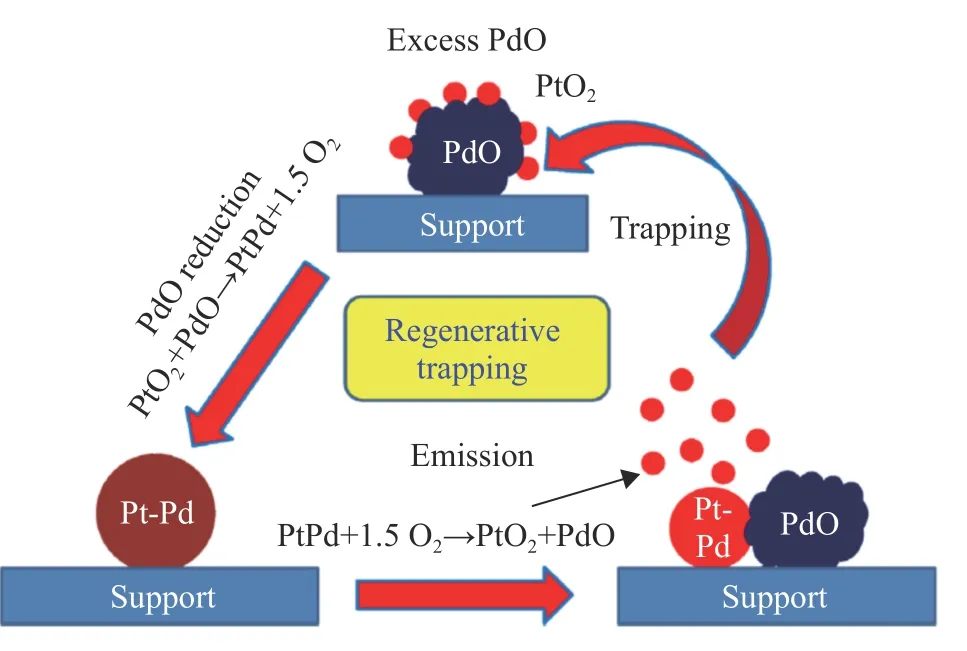
图4 贵金属催化剂再生示意图[36]Fig.4 Schematic showing the regenerative trapping mechanism[36]
2.3 空间限域策略
将金属颗粒置于有限空间内,不仅可有效提高贵金属抗烧结性能,还能够使得贵金属纳米颗粒在有限空间内展现出特异的纳米尺寸效应,改变贵金属d 轨道电子结构特性影响催化活性.如采用对贵金属颗粒生长具有一定限制作用的介孔或者微孔材料.Ghosh等[37]利用SBA-15、MCM-41和硅凝胶制备的介孔材料探究了孔径、孔道长度对Pt 纳米颗粒增长的影响,研究表明孔径小,孔道长都不利于Pt 发生团聚和生成气态PtO2逃逸至气氛中.Wang等[38]采用一步法制备出PdO@纯硅分子筛Silicalite-1,PdO 颗粒存在于有限的空间并且外层纯硅分子筛阻挡了水分子进入,使其700 ℃水热老化后仍保持稳定的活性,当升温至800 ℃后,PdO 颗粒才开始有团聚现象产生.多孔氧化硅材料一定程度上延缓了贵金属烧结现象,但是不能够完全避免,这是由于二氧化硅与贵金属间结合较弱.此外,将贵金属颗粒夹在中间形成三明治结构的制备方法同样能够防止贵金属团聚.Ament等[39]采用氢离子交换的层状钛酸盐将Pd 颗粒夹在中间的三明治结构使得载体与Pd 作用增强了热稳定性和催化活性(图5).Lu等[40]先将Pd 颗粒负载于氧化铝载体上,之后采用原子层沉积法覆盖几个纳米厚度的氧化铝层,在675 ℃运行28 h后分布于新鲜催化剂上的Pd 粒径为2.8 nm,表面未覆盖氧化铝层催化剂Pd 颗粒增大为4.6 nm.Kothari等[41]以铂酸钡(Ba3Pt2O7)为前驱体,利用溶出法将15 nm 的Pt 颗粒嵌入钙钛矿表面,在800 ℃空气中老化350 h 后依然保持15 nm 的Pt 纳米颗粒尺寸.
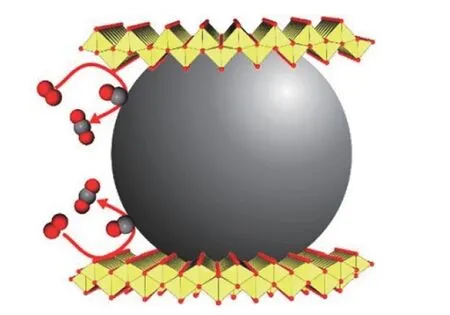
图5 贵金属空间限域示意图[39]Fig.5 Schematic showing the confinement of precious metal nanoparticles[39]
2.4 调控贵金属−载体相互作用
贵金属和载体间相互作用的强度,即金属载体强相互作用(Strong metal-support interaction,SMSI),决定催化剂在高温、高湿度下能够表现出的催化活性[42].SMSI 作用使得贵金属与载体界面间电荷转移、贵金属d 轨道电子特性发生变化,进而影响其催化活性,此外这种相互作用使得载体与贵金属间的作用力增强在高温和高湿气氛下仍保持结构稳定[43].当提高贵金属或γ-Al2O3的热稳定性时,会不可避免对SMSI 作用产生影响进而降低或提高催化剂活性.本小节对在提高催化剂热稳定性的同时不降低催化剂活性的策略进行介绍和讨论.
2.4.1 提高表面五配位铝(Al3+penta)物种
贵金属与载体的SMSI 作用与载体表面电子缺陷直接相关,在载体表面构筑丰富电子缺陷可提高γ-Al2O3催化性能.Kwak等[44]利用超高磁场固体核磁共振图谱(NMR)和密度泛函理论计算(DFT)相结合发现,Pt 稳定性与载体上Al3+penta直接相关.如图6 所示,27Al-NMR 在13×10–6和70×10–6位置的信号峰为八配位Al3+octa和四配位Al3+tetra,在35×10–6处的信号峰为Al3+penta;当负载Pt 之后Al3+penta的峰强度明显下降.这表明,Pt 能够锚定在具有不饱和配位的Al 位点上,这一推论也通过高分辨扫描透射显微镜(STEM)观察到原子态分布的Pt 原子得以验证.当表面Pt 负载质量分数低于1%时,每个Pt 原子单独占据一个Al3+penta位点;当Pt 数量超过γ-Al2O3表面Al3+penta数量,PtO以二维片状锚定在Al3+penta位点周围.这种以单原子或原子簇形式存在的Pt 与载体间强相互作用确保Pt 原子在400 ℃仍保持分散状态而不发生烧结现象.
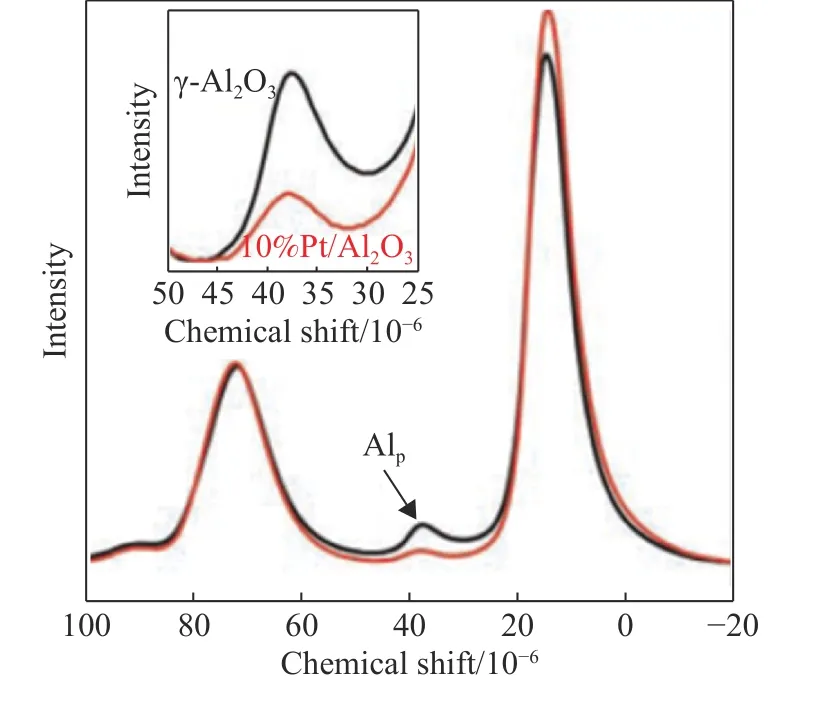
图6 负载Pt 前后γ-Al2O3 上Al 物种数量变化的27Al MAS-NMR 图谱[44]Fig.627Al MAS-NMR spectra of γ-Al2O3 (black) and 10% Pt/γ-Al2O3(red)[44]
如何提高载体中Al3+penta的数量仍然是在催化剂制备中需要研究和探讨的问题.Wang等[45–46]以乙酰丙酮铝和正硅酸乙酯为前驱体,利用火焰燃烧法在约1800 ℃制备出无定型硅铝氧化物,将Si元素引入氧化铝体系内提高了Al3+penta的含量并通过调节硅铝比实现了对Al3+penta含量的控制.当Al 的物质的量分数(Al/[Al+Si])从5%提升至70%,Al3+penta占总Al 种类数量比例从0 升高至55.5%,材料经过脱水处理后Al3+penta占比可进一步提高.虽然火焰燃烧法为我们提供了制备思路,但是在粉体制备过程中需要使用大量有机液体作为助燃剂和分散剂以提高火焰温度和控制粉体粒径,这极大地提高了生产成本.此外,在制备过程中产生大量细微粉体透过玻璃纤维滤膜逃逸造成产率降低,这些因素限制了该方法的应用.因此,仍然需要探索反应条件温和、可批量化生产的制备方法或工艺.
2.4.2 界面重铸法
贵金属与γ-Al2O3间相互作用弱于CeO2和TiO2,即使贵金属能够与Al3+penta结合提高热稳定性,但是当温度高于450 ℃仍会出现烧结现象[44].在γ-Al2O3表面引入其他元素提高贵金属与载体间结合力是改变这一现象的有效办法.如在正硅酸乙酯体表面进行修饰形成几个纳米厚度的SiO2层,可明显提高Pd 分散度,并提高了活性氧物种数量使得甲烷达到90%转化率的温度T90降低了50 ℃[47–48].Zhan等[49]将Mg 掺杂入氧化铝体相内提高了载体热稳定性,优化Mg/Al 比例为1∶3时,Pd→PdO 转化能力最好,表现出最优甲烷催化氧化活性,相比无Mg 掺杂的γ-Al2O3,T90降低了150 ℃.CeO2掺杂不仅可以作为γ-Al2O3的结构稳定剂,亦可以提高贵金属与载体间相互作用力,CeO2表面丰富的活性氧物种与贵金属间以Ce–O–M 结合防止贵金属团聚增强载体热稳定性[50].如图7 所示,Jeong等[51]采用湿法浸渍将CeO2以纳米岛形式锚定在γ-Al2O3,贵金属以单原子形态分布于CeO2纳米岛上;此催化剂在质量空速为2×105mL·h–1·g–1时,150 ℃下实现丙烯和一氧化碳完全转化为水和二氧化碳,275 ℃下丙烷达到完全降解,经过900 ℃水热老化24 h 后活性仍得到保持,表现出优异的活性和热稳定性.
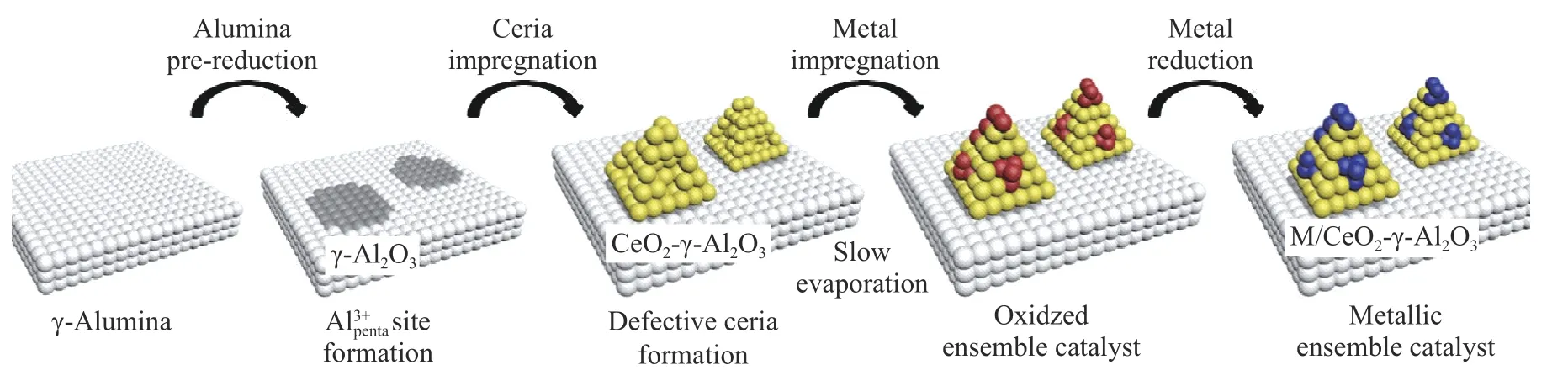
图7 界面重铸法催化剂制备示意图[51]Fig.7 Schematic showing the preparation of metal ensemble catalysts[51]
3 不同抗烧结策略对催化活性的影响
催化剂的稳定性和活性往往同时受到一种或多种因素的影响和制约,在进行催化剂设计和调控时应兼顾稳定性和活性.Cargnello等[52]探究了不同抗烧结策略组合对催化剂稳定性和活性的影响,如图8 所示.图8(a)为多孔CeO2作为壳层包覆在Pd 颗粒上(Pd@CeO2)保证了Pd 颗粒在高温下的稳定,将其负载于以三乙氧基(辛基)硅烷修饰的γ-Al2O3表面确保Pd@CeO2的均匀分散,测试表明在350 ℃下对甲烷降解接近100%,经过850 ℃热处理后仍具有稳定的活性;图8(b)和8(c)表示Pd负载于CeO2或表面有CeO2的γ-Al2O3上分别在接近600 ℃和650 ℃时甲烷完全降解.这表明,相比CeO2,将CeO2转移至γ-Al2O3不但提高Pd 抗烧结能力亦可对催化活性产生积极影响.
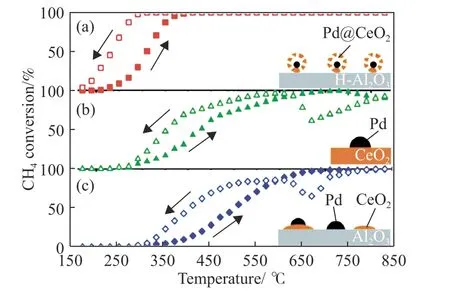
图8 三种不同结构催化剂在10 ℃·min–1 的升降温过程中甲烷催化氧化效率与温度关系.(a) Pd@CeO2/疏水改性的γ-Al2O3;(b)Pd/CeO2;(c)Pd/CeO2/Al2O3[52]Fig.8 Light-off curves of CH4 conversion against the temperature for the three catalysts formulations used at heating and cooling of 10 ℃·min–1: (a) Pd@CeO2/H-Al2O3 core-shell catalyst;(b) Pd/CeO2-IWI;(c) Pd/CeO2/Al2O3-IMP[52]
如何同时提高催化剂稳定性和活性是催化剂制备过程中值得关注和探究的议题.Velinova等[53]在γ-Al2O3中引入La 和Pd 生成稳定相La2PdO4,再引入Ce 利用Ce4+→Ce3+价态变化促进Pd0→Pd2+的变化改善催化剂活性.Wu等[54]也做了相似研究,将Ce、Ni、Co 和Mg 作为活性助剂,Zr 作为提高稳定性的助剂用于甲烷催化降解.Lee等[55]将Pt 和Pd 分步浸渍制备出非均匀相Pt–Pd 合金提高了催化剂使用寿命和甲烷转化温度.Cai等[56]通过制备暴露高能晶面的γ-Al2O3负载Pd 亦能达到如此效果.表2 对负载贵金属的γ-Al2O3催化剂的不同抗烧结策略及其对性能的影响进行了梳理.
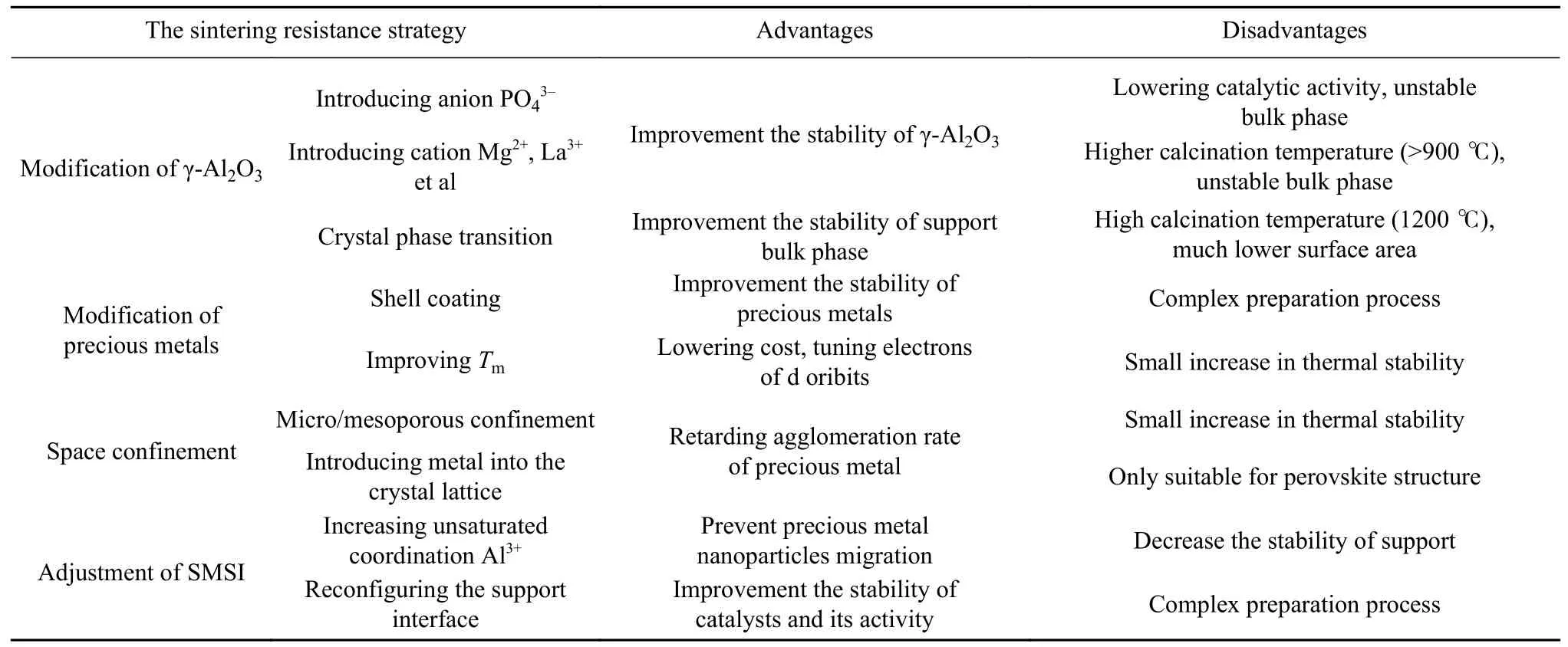
表2 负载贵金属γ-Al2O3 催化剂的不同抗烧结策略Table 2 The sintering resistance strategy of γ-Al2O3 loaded with precious metals
为提高催化剂热稳定性而引入其他变量改变催化剂结构、性质等不可避免会影响催化剂活性.相比其他策略,在γ-Al2O3界面引入阳离子和界面重铸法能够兼顾催化剂活性和稳定性.以在γ-Al2O3界面引入La 为例,La 与界面上Al 原子形成LaAlO3钙钛矿结构[24],贵金属Pd 或Pt 能够嵌入其晶格内,以LaxPd(Pt)yAlO3或者LaPd(Pt)yAlxO3(x+y=1)形式存在[53],当界面上发生还原反应时,界面上的晶格氧容易被夺取,Pd 或Pt 离子从晶格内溢出形成金属态,而钙钛矿结构的破缺导致气相中氧原子吸附,金属态贵金属被氧化,如此往复改善了贵金属的分散度和活性;界面构筑法通常在γ-Al2O3界面引入CeO2,利用Ce4+→Ce3+价态变化促进Pd/Pt0→Pd/Pt2+这一变化提高催化剂活性,此外贵金属与CeO2的SMSI 作用更强确保了贵金属高温稳定性[51].
4 结语与展望
贵金属基催化剂是最具广普性的活性成分.Al 相比其他金属在地球上丰度更高且价格更低具有成本优势,而γ-Al2O3作为载体具有比表面积高、界面酸性位点丰富、可调变性强,且相比其他金属氧化物而言,对气氛中S、N、Cl 等元素表现出更好的耐受性.此外,γ-Al2O3为过渡晶相,在高温下会逐渐发生晶型转变最终形成α-Al2O3.虽然α-Al2O3可耐受1000 ℃以上高温且具有更好的机械稳定性,但是α-Al2O3与贵金属间作用力较弱,且界面上存在较少的缺陷位点,导致贵金属在较低温度下就很容易发生团聚.因此,负载贵金属的γ-Al2O3作为VOCs 催化燃烧技术中的催化剂仍具有不可替代的地位.为确保催化剂高温耐受性,可采用以下策略:引入其他化学元素对γ-Al2O3表面改性提高载体热稳定性和载体与贵金属间相互作用;对贵金属组分进行结构设计(核壳结构、空间限域等)抑制贵金属团聚.
近年来,熵驱动策略催化剂设计的概念逐渐受到广泛的关注.在多组分高熵氧化物(HEOs)体系的热稳定性取决于吉布斯自由能(G=H–TS),而TS增量远大于H增量使得体系具有良好的热稳定性.高熵合金无论作为载体抑或活性组分均能够展现优异的活性和热稳定性,将贵金属引入HEOs 体系内会形成一系列性能优异的催化剂新材料是未来的发展方向.
猜你喜欢
证券市场周刊(2024年13期)2024-04-16 04:33:35
上海金属(2021年6期)2021-12-02 10:47:20
贵金属(2021年1期)2021-07-26 00:39:20
贵金属(2021年1期)2021-07-26 00:39:20
昆明医科大学学报(2021年3期)2021-07-22 07:40:04
——庆祝中国共产党成立一百周年贵金属纪念币展
中国钱币(2021年4期)2021-02-26 00:58:18
生物学通报(2019年3期)2019-02-17 18:03:58
中国塑料(2016年7期)2016-04-16 05:25:52
华东理工大学学报(自然科学版)(2015年4期)2015-12-01 04:00:36
合成材料老化与应用(2015年4期)2015-07-25 10:45:44
