
离子液体型表面活性剂的胶束电动色谱法手性拆分4种酸性药物
2023-01-19 06:16杨轲,于涛
中国药科大学学报 2022年6期
杨 轲,于 涛
(中国药科大学理学院,南京 210098)
手性药物的对映异构体往往表现出不同的生物活性和不良反应,部分对映异构体对人体及环境伤害巨大。自“反应停”等诸多重大事件后,关于手性药物毒副作用的认识及高效简便的识别检测技术已成为药物分析专业领域研究及教学的重要内容[1],但由于对映体之间理化性质几乎相同,常规分离分析手段很难实现分离。近年来,毛细管电泳(CE)对于手性药物分离表现出极大的优越性[2]。其中,由Terabe等[3]在1984年提出的胶束电动毛细管色谱(MEKC),更是将分析物的范围从带电物质扩展到了中性化合物,使得CE 成为最有效的手性分离技术之一。
在MEKC 模式下,溶质不仅可以因为表观淌度差异而分离,还可以基于在水相和在电场中的胶束相之间的分配系数不同而被分离,从而使在毛细管区带电泳(CZE)模式下不能被分离的中性分子得到分离。因此,MEKC的分离能力很大程度上依赖于形成胶束的表面活性剂的结构[4]。表面活性剂按亲水端基团的带电性可以分为阴离子型、阳离子型、两性离子型和非离子型,其中,阴离子型表面活性剂SDS 是最常用于手性分离体系的表面活性剂[5-6]。
离子液体是一类在室温附近或低于100 ℃呈现液态状态的盐类的统称,具有较低的熔点、可忽略的蒸汽压、高的热稳定性、良好的溶解性和环境友好等特点,被广泛应用于多个领域[7-9]。具有长烷基链的离子液体具有明显的两亲性,被称为表面活性离子液体,是一种具有离子液体和表面活性剂结合性质的功能性离子液体[10]。近几年的研究表明,新型的离子液体表面活性剂表现出很好的作为MEKC 体系准固定相的潜力[11-15]。已报道的离子液体表面活性剂以阳离子表面活性剂居多,即其长烷基链连接在离子液体的阳离子部分,而阴离子型离子液体型表面活性剂用于MEKC 的研究少见报道,特别是用于手性分离的研究几乎没有。
本研究依据文献[10],用类似的方法合成了阴离子型离子液体表面活性剂N-丁基-N-甲基吡咯烷十二烷基硫酸盐([C4MP][C12SO4])(结构式见图1),并将其与中性手性选择剂葡萄糖基-β-环糊精(Glu-β-CD)构建CD-MEKC 体系在低pH 条件下分离4种酸性手性药物(萘普生、华法令、酮洛芬和布洛芬,结构式见图2)。
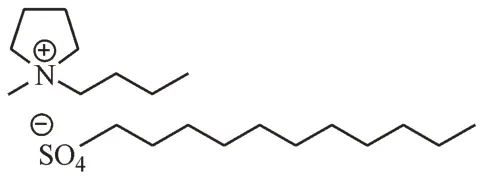
Figure 1 Stucture of [C4MP][C12SO4]
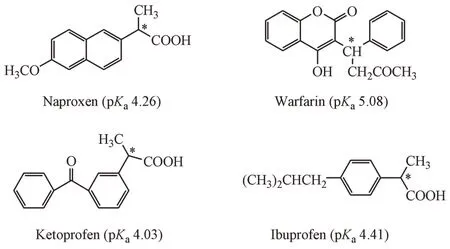
Figure 2 Chemical structure of naproxen,warfarin,ketoprofen and ibuprofen
1 材 料
1.1 试 剂
N-丁基-N-甲基吡咯烷氯盐([C4MP]Cl,上海成捷化学有限公司);Glu-β-CD(≥ 98%,山东淄博千汇精细化工有限公司);萘普生和华法令(四川西亚试剂有限公司);酮洛芬和布洛芬(美国Sigma-Aldrich 公司)。所有的手性药物都是消旋体混合物。甲醇、乙醇、乙腈和异丙醇(色谱纯,江苏汉邦科技有限公司);磷酸二氢钠(≥ 99.0%)、磷酸(上海凌峰化学试剂有限公司);实验用水为二次蒸馏水。
1.2 仪 器
Agilent3DCE G7100A(美国Agilent 科技公司);未涂层熔融石英毛细管(河北永年锐沣色谱器件有限公司);BT-25S 电子天平和PB-10 型pH 计(德国Sartorius 公司);N-1100D-WD 旋转蒸发仪(日本Eyela公司)。
2 方 法
2.1 电泳条件
未涂层石英毛细管柱(33 cm × 50 µm,有效长度24.5 cm)。运行缓冲液为pH 3.0(萘普生、华法令、酮洛芬)、pH 3.3(布洛芬)的50 mmol/L磷 酸 盐(含30 mmol/L Glu-β-CD、40 mmol/L [C4MP][C12SO4]和20%甲醇);运行电压:-15 kV或-20 kV;检测波长:萘普生(235 nm)、华法林(210 nm)、酮洛芬和布洛芬(225 nm);压力进样,30 mbar(1 bar = 6.895 kPa),4 s;运行温度,25 ℃。
2.2 溶液的配制
2.2.1 运行缓冲液配制 缓冲盐溶液为50 mmol/L磷酸缓冲盐(含有一定量的有机溶剂)。运行缓冲盐全部当时配制,将Glu-β-CD和阴离子型表面活性剂溶于包含有机溶剂的缓冲盐溶液(特定pH)中,用极少量的10%磷酸溶液调节至所需的pH。
2.2.2 样品溶液配制 分别称取萘普生、华法令、酮洛芬、布洛芬适量,用水-甲醇(1∶1)溶解,进样质量浓度均为0.5 mg/mL。
所有样品溶液及运行缓冲溶液经0.45 µm 微孔滤膜过滤后使用。
2.3 离子液体表面活性剂[C4MP][C12SO4]的合成
[C4MP][C12SO4]的合成参照文献[10],具体步骤如下:将[C4MP]Cl和SDS 取相同当量,置圆底烧瓶内,加入适量的二氯甲烷,室温下搅拌4 h。抽滤,去除沉淀物,有机相用水洗数次,用旋转蒸发仪去除有机溶剂,真空干燥24 h,得蜡状离子液体。
3 结果与讨论
3.1 手性分离体系的构建
[C4MP][C12SO4]作为一种新型离子液体表面活性剂,在298 K下的水溶液中的临界胶束浓度(CMC)为2.7 mmol/L,同条件下SDS的CMC为8.1 mmol/L。[C4MP][C12SO4]拥有优越的表面活性,且由于其阳离子为吡咯烷阳离子,紫外吸收极弱,作为背景添加剂,对样品干扰少,基线平稳。本研究使用的表面活性剂浓度均在其CMC以上。
实验中首先考察了CZE 模式和MEKC 模式下,Glu-β-CD手性选择剂对4种酸性药物(萘普生、华法令、酮洛芬和布洛芬)的拆分效果。结果表明,在低pH 条件的CZE 下,酸性药物均不带电,中性环糊精不能实现对4种药物的手性分离;当缓冲液pH大于药物的pKa时,萘普生和华法令可实现部分分离,酮洛芬和布洛芬不能被分离。因此,本研究建立低pH 条件下的MEKC 体系,低pH 条件下,酸性药物呈电中性,药物根据其在含环糊精的缓冲液相中和胶束相中的分配不同而实现分离,环糊精对手性药物的识别效果较好。如采用正电压模式,由于胶束带负电向正极迁移,样品不能出峰,而采用反向电压,样品在带负电的胶束带动下出峰。因此采用负电压模式构建CD-MEKC分离体系。
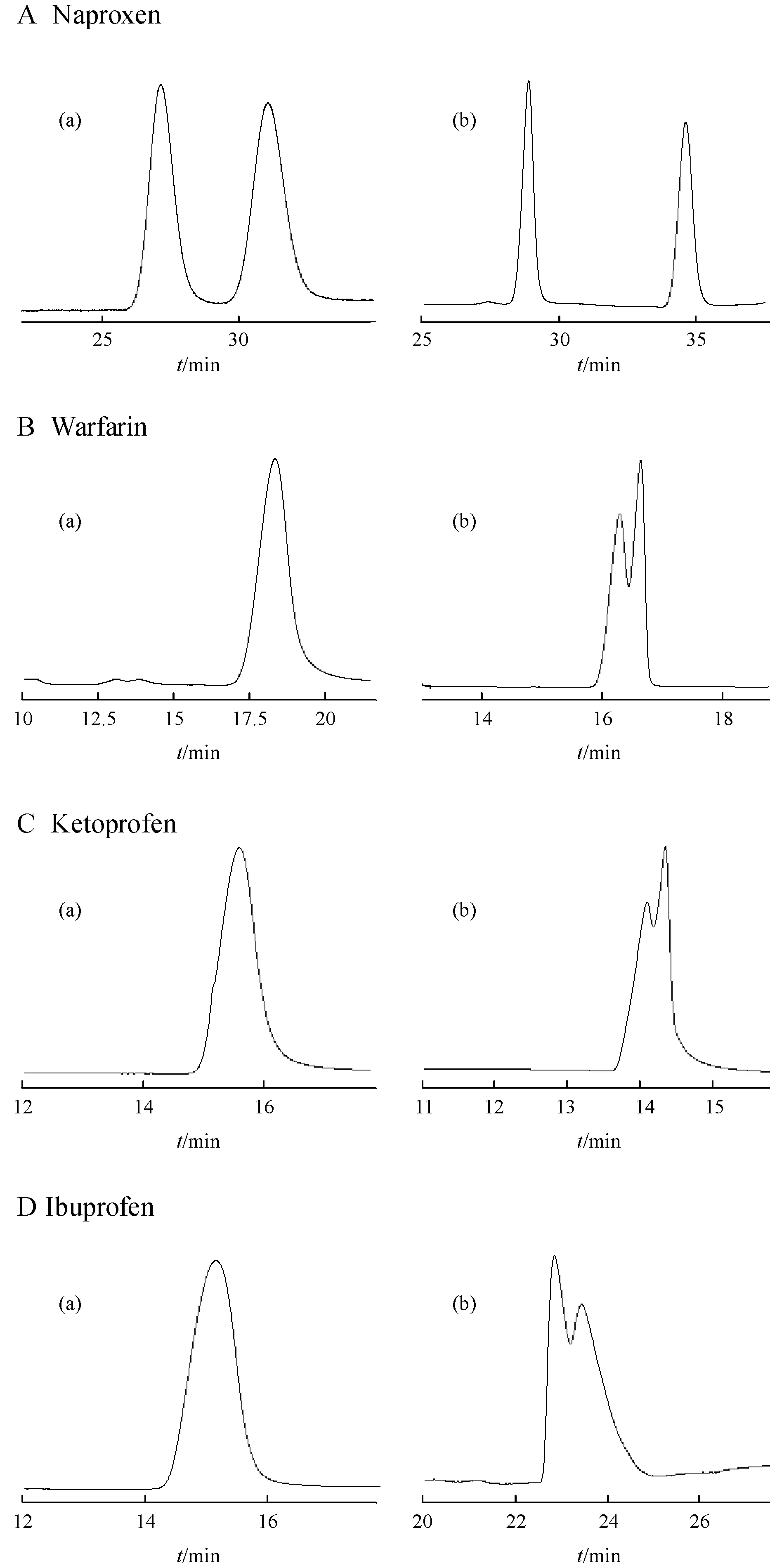
Figure 3 Typical electropherograms of micellar electrokinetic chromatography( MEKC) systems based on SDS and [C4MP] [C12SO4]Conditions: fused-silica capillary, 33 cm (24.5 cm effective length) × 50 µm; 50 mmol/L phosphate buffer (20% of methanol included) containing 30 mmol/L Glu-β-CD and (a) 40 mmol/L SDS; (b) 40 mmol/L [C4MP][C12SO4]; (A) and (B) pH 3.0, applied voltage -15 kV; (C) pH 3.0, applied voltage -20 kV and (D) pH 3.3,applied voltage -20 kV; capillary temperature, 25 °C
在MEKC 模式下,分别采用SDS 和[C4MP][C12SO4]作为表面活性剂,其中基于SDS 的体系除了可对萘普生实现分离,其余药物无法分离,而基于[C4MP][C12SO4]的体系能大大提高萘普生的分离结果,而对华法令、酮洛芬和布洛芬均可实现部分分离,MEKC体系电泳对比如图3。由电泳图谱可看出,基于[C4MP][C12SO4]的CD-MEKC 体系相比于基于SDS 的体系,对萘普生的分离效果大大改善,其中选择性因子(α)从1.14提高到1.20,分离度从2.05 提高到6.75;对华法令、酮洛芬和布洛芬则从不能分离到部分分离。这可能是由于[C4MP][C12SO4]和SDS 在水相中的聚集行为以及胶束排列都有所不同,有研究表明[C4MP][C12SO4]具有比传统表面活性剂SDS 更优的胶束特征[9]。
3.2 电泳因素对分离的影响
3.2.1 有机溶剂的种类和比例对分离的影响 本研究重点考察了甲醇、乙醇、乙腈和异丙醇这4种有机溶剂对酸性药物分离的影响。在不添加有机溶剂或有机溶剂比例在低于20%时,手性药物在长分析时间(大于60 min)内不能出峰。固定比例为20%,4 种有机溶剂中,甲醇为更适合该CDMEKC 体系的有机溶剂。进一步考察甲醇的比例对分离的影响,发现甲醇比例增大后,手性药物的迁移时间均大大延长,而分离度也呈现下降的趋势,可能是因为迁移时间较长,分离效率下降,峰展宽导致分离结果变差。因此,选择20%甲醇作为该系统的有机溶剂。
3.2.2 缓冲盐浓度及pH 对分离的影响 通常,缓冲盐种类及浓度会影响缓冲液离子强度、电渗流大小及溶质在毛细管壁的吸附等。当缓冲系统浓度增加时,离子强度增大,会使电渗流减小,有利于减少溶质和管壁的相互作用,改善分离;但浓度过大则会使电流过大,焦尔热影响更为严重,峰展宽进而不利于分离。
选择在低pH 范围内有较好缓冲能力的磷酸盐作为缓冲体系,考察了不同磷酸盐浓度(30、50和70 mmol/L)对药物手性分离的影响。结果发现,随着磷酸盐浓度从30 mmol/L 增至50 mmol/L 药物的分离得到改善;而当磷酸盐浓度由50 mmol/L 进一步增至70 mmol/L 时,电流较大,峰展宽,药物的分离度呈下降趋势。因此,选择50 mmol/L 的磷酸盐作为CD-MEKC拆分的缓冲盐体系。
在50 mmol/L 的磷酸盐(包含体积分数20% 的甲醇)缓冲溶液中加入30 mmol/L Glu-β-CD 并在40 mmol/L [C4MP][C12SO4]条件下,考察了不同缓冲溶液pH(2.7、3.0、3.3、3.6 和3.9)对手性药物分离的影响,结果见表1。
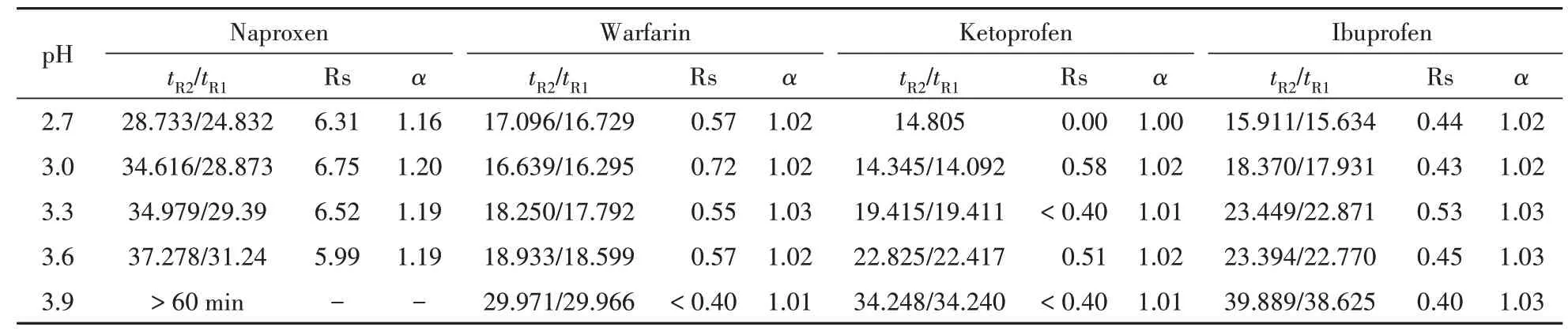
Table 1 Effect of buffer pH on enantioseparation of chiral drugs
表1中显示,在考察的pH 范围内,手性药物的迁移时间随pH 升高而延长。而药物的分离结果在整个pH 范围内变化并不特别明显,可能是因为药物在pH 低于其pKa的缓冲溶液中,解离情况较弱,大部分以分子形式存在,因此,对溶液pH 变化相对不敏感。考虑到分析时间和样品峰形,萘普生、华法令和酮洛芬的最佳pH 为3.0,而布洛芬则选择3.3作为其分离pH。
3.2.3 环糊精浓度对分离的影响 本研究采用
Glu-β-CD 作为手性选择剂建立基于离子液体表面活性剂的CD-MEKC 手性分离体系,为考察环糊精浓度对手性分离的影响,设定其浓度范围为10 ~ 40 mmol/L。考察结果如表2所示。
当环糊精浓度在10 mmol/L 时,除了萘普生有部分分离外,其余手性药物均不能实现分离,说明环糊精浓度过低时不能充分发挥其手性识别能力。当环糊精浓度增大时,可以看出药物的迁移时间延长,分离度和选择性因子(α)提高。而当Glu-β-CD 浓度增大至40 mmol/L 时,手性药物在60 min内均未出峰,可能是由于此时缓冲溶液黏度升高,药物在环糊精中的分配增加,在胶束相中的分配减小,胶束不足以将药物较快带至出口端。考虑到分离度和α达到最佳时,选择30 mmol/L 作为该体系的Glu-β-CD浓度。
3.2.4 离子液体表面活性剂浓度对分离的影响 因为华法令、酮洛芬和布洛芬的分离度在较低的水平,故选择萘普生这一手性药物考察离子液体表面活性剂[C4MP][C12SO4]的浓度,考察范围为35 ~ 60 mmol/L,实验结果见表3。
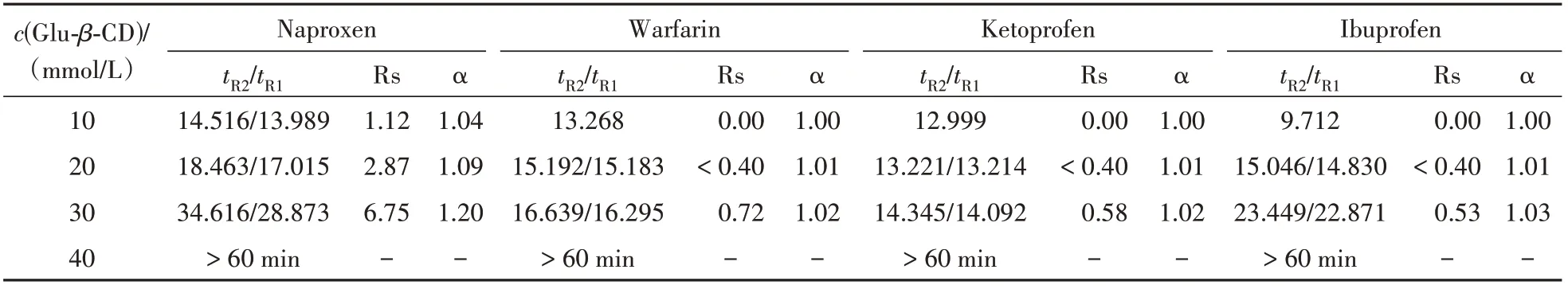
Table 2 Effect of concentration of Glu-β-CD on enantioseparation of chiral drugs
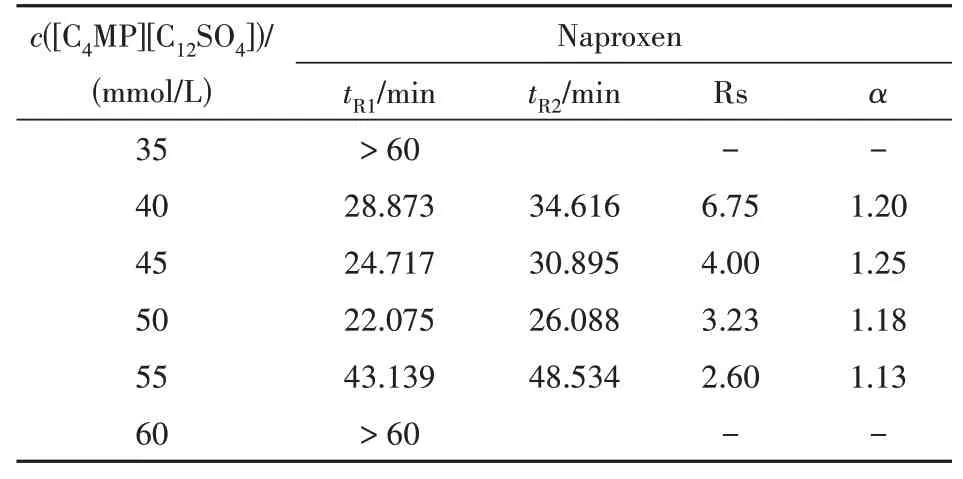
Table 3 Effect of concentration of [C4MP][C12SO4] on enantioseparation of chiral drugs
在考察的浓度范围内,当离子液体[C4MP][C12SO4]浓度低于40 mmol/L 时,萘普生样品峰在较长分析时间内仍未可见。推测可能是因为形成的胶束相浓度太低,在负压模式下胶束无法快速地将萘普生带至检测端。随着[C4MP][C12SO4]浓度升至40 mmol/L时,样品能够出峰,且分离效果极佳。当浓度进一步增大到45 ~ 50 mmol/L,萘普生迁移时间在逐渐缩短,分离度也在降低,可能是因为胶束浓度增大,反而对对映体的区分作用减弱导致的。随着离子液体浓度上升至55 ~ 60 mmol/L,样品的迁移时间突然大大延长,可能是由于阳离子浓度也在增大,使得对毛细管壁的吸附增强,对电渗流产生影响所致。另外则可能是阳离子大量存在中和了负电性胶束表面部分负电荷使胶束粒子表面电荷密度变小,胶束粒子变大,从而影响了胶束相的迁移速度。
3.2.5 运行电压对分离的影响 通常来说,较高的运行电压往往能带来较高的分离效能和较短的分析时间,但是高的电压也会产生过多的焦耳热从而降低分离效率。为了获得合适的运行电压,本研究考察的电压范围为-5 ~ -25 kV,结果表明,降低反向电压的大小,药物的迁移时间延长,如预期的一样,较低的电压能延长分析时间,给予分析物和手性选择剂之间更多的作用机会,然而也会造成分离效率较低和峰展宽等问题,使得样品的分离度随着电压降低呈现先增大后减小的趋势。对于萘普生和华法令来说,-15 kV 为其最佳运行电压;对于酮洛芬和布洛芬,-20 kV 的运行电压则更适合。
4 结 论
本工作合成了阴离子型离子液体表面活性剂[C4MP][C12SO4],并将其与中性手性选择剂Glu-β-CD 用于胶束电动毛细管色谱手性分离体系的构建,首次在低pH 条件下分离4 种酸性药物(萘普生、华法令、酮洛芬和布洛芬)。相比于同条件下的SDS 体系,基于[C4MP][C12SO4]的MEKC 体系大大提高了萘普生的分离效果,并使其余3 种在SDS体系中无法分离的药物实现部分分离。这一研究扩展了Glu-β-CD 在CE 手性拆分中的应用范围,探索了离子液体表面活性剂在MEKC 体系中的分离潜力。同时本团队以文章内容设计“药学拔尖学生培养计划”示教实验,结合健康中国战略,阐述了手性药物研发风险及最新研究进展。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
家庭医药(2019年7期)2019-07-25
家庭医药(2019年13期)2019-01-15
保健与生活(2018年13期)2018-01-26
中国药房(2017年18期)2017-07-25
中国科技术语(2016年3期)2016-12-04
中国癌症杂志(2015年4期)2015-12-09
中国当代医药(2015年8期)2015-03-01
中国卫生标准管理(2015年3期)2015-01-27
中国药业(2014年24期)2014-05-26
