
金属镓在Bi(111)表面上的吸附和合金化①
2023-01-17 07:19卢尧臣张玉峰白萌萌李佐曹龙江陶敏龙
西南师范大学学报(自然科学版) 2023年1期
卢尧臣,张玉峰,白萌萌,李佐,曹龙江,陶敏龙
西南大学 物理科学与技术学院, 重庆 400715
合金用途广泛, 金属合金化通常可以提高其机械性能、催化反应和耐腐蚀性等[1-2]. 表面合金是在单一基板上形成的二维(2D)合金层, 由于其结构明确, 可作为双金属表面的模型系统, 因此受到越来越多的科研工作者的关注[3]. 扫描隧道显微镜(scanning tunneling microscope, STM)[4]具有原子分辨率, 可以在原子级别上表征材料的表面形貌. 因此, STM已成为二维合金结构研究的一种重要方法[5].
金属镓作为形成低熔点[6]合金的重要材料之一, 在室温状态时呈液态. 与液态金属汞相比, 镓毒性微弱, 应用领域更为广泛. 金属镓作为GaAs和GaN等材料的重要组成元素[7-8], 在现代半导体工业中有重要的价值. 同时, 镓作为低熔点金属易与铝、铋、铟和锡形成低熔点合金, 其中镓铟合金和镓铟锡三元合金是镓基合金近几十年来的研究重点[9-12]. 应用镓的低熔点这一特性开发出的镓基合金, 如镓铟合金(GaIn, 75%镓, 25%铟, 熔点为15. 7 ℃)和镓铟锡合金(GaInSn, 68%镓, 22%铟, 10%锡, 熔点可达0 ℃以下)[13], 在常温下呈现液态, 在散热、柔韧器件和液态机械领域具有重要的潜在利用价值[14-17].
金属铋属于VA族元素, 具有和金属镓类似的一些特殊性质, 如热缩冷胀等, 同时铋薄膜表面具有很强的量子尺寸效应, 一直是研究的热门材料. 金属镓在液态时易腐蚀其他金属形成合金, 这种腐蚀性更确切的表述是侵润, 只有在镓处于液体状态时才能通过固体表面较小的缝隙进入其内部. 因此, 在微观领域中镓与铋相遇是否会有新的有趣现象或者是否会形成新的合金结构值得进一步探索. 目前, 国内外对镓铋合金的研究只限于高纯度金属镓和金属铋块体固溶后形成固溶体合金的性质和结构, 如Ga0.985Bi0.015[18]和Ga39Bi61[19]固溶后得到的不同掺杂浓度的镓铋合金. 这些研究很少能直接观察单个镓原子沉积在衬底上的吸附、扩散、聚集, 以及镓铋合金的形成过程和结构特点. 基于镓基液态金属的特性, 本研究制备了在Bi(111)表面的镓基合金层, 并通过STM来探究合金形成条件和特性[20], 如吸附、扩散、聚集和镓原子与衬底的合金化过程等. 本实验采用Bi(111)作为衬底[21], 通过控制实验进程, 研究不同衬底温度对镓原子沉积在Bi(111)表面的影响和形成的结构, 然后通过基于密度泛函理论(density functional theory, DFT)的第一性原理计算来进行验证. 当衬底温度较低时(200 K以下), 大部分镓原子在Bi(111)表面发生聚集, 形成镓液滴, 少部分镓原子因沉积时速度较快, 最终迁移到铋岛台阶边缘处, 逐步侵蚀铋原子层, 形成镓铋合金. 当衬底放置到室温后, 经过一段时间, 由镓原子聚集而成的小液滴会消散, 吸附在衬底表面的镓原子侵蚀形成的合金结构区域会增大.
1 实验部分和模拟计算
1.1 实验操作
本研究所采用的设备是日本Unisoku公司生产的超高真空-低温扫描隧道显微镜(LT-STM), 实验本底真空优于1.0×10-9Pa. Si(111)样品在500 ℃下除气8 h, 将其快速升温到1 200 ℃以除去样品表面的氧化膜, 然后缓慢降至室温, 得到干净的Si(111)-7×7表面. 将氮化硼坩埚中的高纯度(99. 98%)铋金属颗粒除气20 min后, 利用热蒸发将其沉积在洁净的Si(111)-7×7表面上, 在大约400 K温度下退火2 h, 得到平整有序的Bi(111)薄膜. 单质镓置于石英坩埚内, 在750 ℃下除气3 h后, 将其沉积在Bi(111)表面上, 再迅速传入STM腔体进行原位检测. 本文中所有的STM图都是在液氮(78 K)温度下, 采用恒流模式采集得到的.
1.2 计算方法
模拟计算基于DFT的第一性原理方法. 计算过程中采用了广义梯度近似(generalized gradient approximation, GGA)下的加密算法(password based encryption, PBE)方法来计算电子之间的交换关联能[22]. 平面波基组构造波函数的截断能为400 eV, 离子和电子的相互作用采用投影缀加平面波方法(projector angmented-wave, PAW)来解释[23-24]. 为了模拟真实的Bi(111)表面, 衬底选取了包含6个铋原子层的4×4超胞模型, 其中底层的两个原子层被固定, 用来模拟块体效应, 沿着衬底的Z方向取1.5 nm的真空层以消除周期性图像带来的影响. 电子性质和力的收敛标准分别为10-6eV和0.1 eV/nm. 在结构弛豫时采用3×3×1的k-mesh进行优化.
2 结果和讨论
首先, 在衬底温度较低时(200 K以下)将镓原子沉积在Bi(111)表面, 传入低温STM腔进行原位检测. 图1a是平整的Bi(111)表面的STM图, 右上角的插图是Bi(111)的原子分辨图. 图1b是在Bi(111)表面沉积镓原子后的STM图. 图1d是镓原子聚集形成的小液滴高度图. 相比于原本平整的Bi(111)表面, 能够明显观察到镓原子聚集在衬底表面形成的直径约为10~30 nm的红色高亮球状液滴, 这些大小不一的红色小球与周围铋层之间的高度差大约为2~10 nm. 除此之外, 从图2b中蓝色圆圈标注区域可以看到, 沉积镓原子后的Bi(111)表面形貌也发生了改变, 在Bi(111)台阶边缘处出现了小块的较暗的新台阶, 高度略低于铋台阶的高度. 将样品在超高真空室温环境下退火8 h后, 样品中绝大部分的液滴会脱附, 被真空泵抽离. 如图1c所示, 原本图像中由镓原子聚集形成的红色高亮的液滴小球已经消失, 仅留下Bi(111)台阶边缘较暗的新台阶.
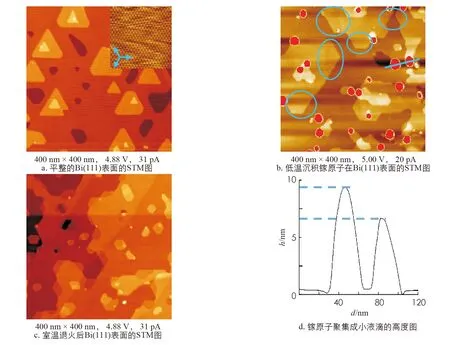
图1 镓原子沉积在Bi(111)表面的STM图
为了进一步探究沉积镓原子后对Bi(111)表面的影响, 研究了样品在放置室温后的Bi(111)表面的少量镓原子沉积区域(图2a). 图中能够观测到原本存在于Bi(111)表面上三角形形貌的铋岛和铋岛台阶边缘处依附于三角形铋岛的小块新台阶. 如图2b所示, 依附于三角形铋岛的新台阶的高度约为0.33 nm, 低于Bi(111)的台阶高度0.08 nm左右. 在三角形铋岛的台阶边缘处, 依附在铋岛台阶边缘的小块台阶区域与原本铋岛之间形成的新台阶向内凹陷, 这是沉积镓原子后Bi(111)台阶被吸附的镓原子所侵蚀形成稳定合金后的形貌. 因此, 本研究称这些沉积镓原子后出现的略低于Bi(111)台阶的合金区域为侵蚀区域. 被镓原子侵蚀后形成的合金区域台阶不与铋台阶接触的边界是较规则的直线, 而与原铋岛交界的台阶是不规则的曲线. 图2c是Bi(111)台阶边缘存在大面积合金的STM图像. 图2c中大面积合金区域与图2a中小块的侵蚀区域相同, 边缘处是规则的直线, 交界处向内凹陷, 大的合金区域应是小的区域逐步扩大连接形成的. 图2d是图2c中蓝色方框标记处侵蚀区域与铋台阶交界的STM高分辨图. 图2d中右半部分为交界处较高的台阶, 是Bi(111)的STM原子分辨图, 而左边较暗的侵蚀区域即为形成的稳定镓铋合金结构的STM原子分辨图. 侵蚀区域的原子排列方式和衬底Bi(111)的排列方式相同, 都是六角密排结构, 原子排列方向也与衬底相同, 但高度略矮于Bi(111)的原子层.

图2 少量镓原子沉积区域Bi(111)表面的STM形貌图和合金区域高分辨STM图
合金形成局限于表面层的现象已在许多其他异质外延系统中得到证实, 包括一些存在大块混相的系统中. 形成稳定结构的原因不仅仅是表面应变能最低, 还应包含体系的系统总能量最低. 基于Bi(111)表面沉积镓原子的实验中出现的镓铋合金结构, 利用第一性原理方法来对镓原子直接吸附在衬底Bi(111)表面的情况和替代铋原子形成合金的情况进行模拟计算, 比较不同吸附位置上镓原子吸附能量和替代不同位置的铋原子形成表面合金的掺杂能量. 如图3a所示, Bi(111)表面不等价的吸附位置有4个, 分别为top(A)、bridge(B)、正对第2层铋原子上方的hcp(C)位和第3层铋原子正上方上fcc(D)位[25]. 镓原子吸附能定义为:
Eads=Etot-EBi(111)-EGa
式中: 这里Etot是Bi(111)表面吸附单个镓原子后的体系总能量,EBi(111)是Bi(111)表面的能量,EGa是单个镓原子的能量. 镓原子替代铋原子形成合金有可能是镓原子替代表层铋原子, 也有可能当镓原子能量较高时直接进入铋内部替代第2层的铋原子, 因此在模拟计算中分别考虑了这两种情况. 如图3b所示, E和F分别表示镓原子替代第1层和第2层铋原子. 镓原子取代铋原子形成稳定合金结构的内聚能量定义为:
Eads=Etot-EBi(111)+EBi-EGa
式中:Etot是铋表面形成镓铋合金后的体系总能量,EBi(111)是Bi(111)表面的能量,EBi是单个Bi原子的能量,EGa是单个镓原子的能量.
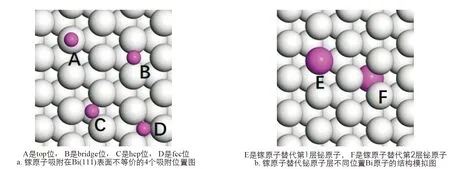
白色原子代表Bi原子, 紫色代表Ga原子
计算后各吸附位置的吸附能和替代铋原子形成合金的内聚能见表1, 其中吸附在bridge和top位置的镓原子计算优化后呈现在hcp位置.

表1 在Bi(111)表面镓原子不同位置的吸附能和内聚能
从表1中数据可以看出, 在Bi(111)表面不等价的吸附位置中镓原子吸附在hcp位置时能量较低, 替代铋原子形成表面合金的能量更低, 第2层铋原子被替代形成的合金结构的能量最低, 掺杂能量为-2.62 eV. 这些数据说明镓原子沉积在Bi(111)表面更易于替代铋原子形成表面合金, 而且替代第2层铋原子形成的合金结构最为稳定. 镓原子替代第2层铋原子形成合金结构时, 吸附在表面的镓原子从铋台阶边缘开始侵蚀铋原子层, 逐步替代第2层铋原子, 最终在第2层铋原子被完全替代的区域形成稳定的合金结构. 图4a和图4b分别是经过第一性原理计算所得的镓铋合金结构的俯视图和侧视图. 俯视图是镓原子完全取代第2层铋原子形成的镓铋合金结构, Bi(111)面的原子排列方式与形成的镓铋合金结构的原子排列方式都为六角密排结构, 合金结构的晶向也与衬底相同. 从侧视图中可以看到相比于原本铋的双原子层之间的距离镓铋合金结构的镓原子层与上层铋原子层的更加接近, 替代铋原子后形成的合金层高度相较于原本的铋原子层高度略矮. 实验所得STM图中Bi(111)具有双层结构[26], 它的原子层厚度约为0.42 nm, 而形成的镓铋合金结构的原子层厚度为0.33 nm(图2b), 这与模拟所得的镓铋合金高度0.33 nm(图4b)结果一致. 当衬底温度较低时(200 K以下), 部分镓原子沉积时动能较小, 聚集在衬底表面形成液滴小球, 部分镓原子进入衬底替代第2层铋原子形成稳定合金结构; 当样品长时间处在室温的超高真空环境时, 镓原子聚集成的小液滴会在脱附后被真空泵抽离, 仅剩下稳定的镓铋合金.

图4 镓铋合金的第一性原理计算结构图
3 结论
本实验利用超高真空-低温隧道显微镜技术研究了镓原子在Bi(111)表面的吸附和合金化过程, 并结合基于DFT的第一性原理方法进行模拟计算. 镓原子沉积在Bi(111)表面后, 一部分聚集形成大小各异的小液滴, 另一部分与衬底结合形成镓铋合金. 样品在室温下退火8 h后, 镓液滴脱附, 仅留下镓铋合金. 合金层的高度低于Bi(111)台阶高度约0.08 nm, 结构为六角密排结构. 理论模拟结果显示沉积的镓原子会取代第2层铋原子形成镓铋合金, 合金层的厚度为0.33 nm, 与实验结果吻合很好.
猜你喜欢
量子电子学报(2022年4期)2022-08-22
力学学报(2022年2期)2022-03-20
生物工程学报(2020年7期)2020-07-29
中南大学学报(自然科学版)(2019年7期)2019-08-13
中国宝玉石(2018年3期)2018-07-09
劳动保护(2018年5期)2018-06-05
数学大王·低年级(2017年9期)2017-09-18
中南大学学报(自然科学版)(2016年2期)2017-01-19
中国科技信息(2015年24期)2015-11-07
中国科技信息(2015年23期)2015-11-07
