
基于公共数据库分析自噬相关基因在肺腺癌患者中的预后意义
2023-01-16 04:28王玉然王文浩
医学信息 2022年22期
李 敖,王玉然,王文浩
(潍坊医学院附属医院肿瘤科1,放疗科2,山东 潍坊 261041)
截至2020 年,肺癌发病率位居第2 位,死亡率高居首位[1,2]。肺腺癌是肺癌常见的组织学亚型之一。肺癌发病机制、进展及耐药的分子机制是肺癌精确治疗的研究热点,也是药物治疗的关键。可靠的生物标志物对于肺癌的准确诊断和治疗至关重要。研究发现[3],自噬在非小细胞肺癌的发展、治疗和耐药中起重要作用。自噬是发生在特定生物过程中,由多种信号通路调控的一种细胞程序性死亡方式[4],其可参与肿瘤生长调控[5,6]。在肺癌细胞学实验中,表皮生长因子受体酪氨酸激酶抑制剂可以激活自噬。本研究利用生物信息学方法对肺腺癌中自噬相关基因(autophagy-related genes,ATGs)的差异表达进行分析,根据Cox 分析建立ATGs 的预后模型,以期为ATGs 在肺腺癌患者预后评估和靶向治疗中的应用提供新的研究方向,现报道如下。
1 资料与方法
1.1 数据来源 肺腺癌基因表达数据及相关临床基础数据来源于TCGA 数据库(https://portal.gdc.cancer.gov/)。从TCGA 数据库获取54 例正常组织和497 例肿瘤组织的RNA 测序数据,并获取相应的临床数据。利用R 软件对样本中的差异表达基因进行Wilcox 检验。从人类自噬数据库HADb(http://www.autophagy.lu/)中共检索到232 个ATGs。为了筛选差异表达的ATGs,阈值设定为logFC>1.0,FDR<0.05。
1.2 GO 和KEGG 对差异表达ATGs 富集分析 使用R 软件进行GO 富集分析和KEGG 富集分析,其中GO 富集分析包括生物过程(biological process,BP)、细胞成分(cell components,CC)和分子功能(molecular functions,MF)3 类。使用公共蛋白质相互作用数据库PPI(https://string-db.org/)生成差异ATGs 的蛋白质相互作用网络,使用Cytoscape 软件(3.7.2 版)编辑蛋白质相互作用图像。
1.3 预后模型构建 应用R 软件进行单因素Cox 分析,筛选出与肺腺癌预后相关的差异表达基因。利用多变量Cox 回归分析构建肺腺癌ATGs 的预后模型。使用特定的公式计算风险评分,用中位风险阈值将肺腺癌患者分为高危和低危2 组。应用Kaplan-Meier 分析风险评分与肺腺癌患者预后的关系。
1.4 统计学方法 所有统计分析采用R 软件(4.0.4 版)进行。采用对数秩检验和K-M 分析法绘制生存曲线。单因素和多因素分析采用Cox 比例风险回归模型。用双侧t检验进行风险评分与临床参数的统计比较。以P<0.05 表示差异有统计学意义。
2 结果
2.1 ATGs 的差异表达分析 以TCGA 数据库中54例正常组织和497 例肺腺癌组织的mRNA 阵列分析为基础,以绝对mRNA 基因表达水平logFC>1.0 和FDR<0.05 为筛选标准进行ATGs 的差异表达分析。R 软件包“limma”和“ggpubr”分析共获得200 个ATGs。ATGs 分布散点图见图1A。以LogFC>1.0 和P<0.05 为选择标准,筛选出在肺腺癌数据库中差异表达的30 个ATGs,见图1B、1C。

图1 ATGs 在肺腺癌TCGA 数据库中的差异表达
2.2 ATGs 的功能富集分析 在生物学过程中,GO 最显著富集的前3 个功能是内源性凋亡信号通路、巨噬细胞自噬和神经元死亡,细胞成分包括自噬小体、自噬体膜和内质网伴侣复合体。在分子功能上,ATGs 主要是浓缩蛋白磷酸酶结合、磷酸酶结合和蛋白二硫键异构酶活性。KEGG 通路富集分析显示,这些基因与自噬(动物)通路、ErbB 信号通路和IL-17信号通路相关。此外,从在线STRING 数据库中获得了30 个ATGs 的蛋白互作网络,见图2。

图2 GO 和KEGG 富集功能分析与PPI 网络构建
2.3 自噬相关危险因素与肺腺癌预后关系的研究对200 个ATGs 进行单因素Cox 回归分析,从肺腺癌TCGA 数据库(图3A)中获得了21 个预后相关ATGs,其中9 个ATGs(ATG4A、NLRC4、PRKCD、DAPK2、SIRT2、CCR2、ATG16L2、DLC1、DRAM1)被认为是保护性基因(HR<1),其余12 个ATGs(ITGB4、BIRC5、CTSL、SPHK1、APOL1、ITGA6、ITGB1、GAPDH、ERO1A、EIF2S1、MBTPS2、ST13)被认为是危险性基因(HR>1)。通过多因素Cox 回归分析,从21 个ATGs 中筛选出与肺腺癌患者预后相关的8个关键基因(ATG4A、CCR2、MBTPS2、APOL1、ERO1A、SPHK1、ST13 和ITGA6),见表1。通过多因素Cox 回归分析得到了各危险基因的系数值,根据ATGs 公式构建自噬预后模型:风险评分=(-0.5562×ATG4A表达值)+(-0.3777×CCR2 表达值)+(0.3398×MBTPS2)+(0.1319 ×APOL1 表达值)+(0.2030 ×ERO1A 表达值)+(0.1832 ×SPHK1 表达值)+(0.2893×ST13 表达值)+(0.1399×ITGA6 表达值)。根据此公式,从TCGA 数据库获得患者的预后风险评分。图3B 显示了肺腺癌患者的风险评分分布,图3C显示了生存时间和风险评分之间的相关性。建立热图分析低风险组和高风险组中包含的8 个ATGs 的表达差异(图3D),结果显示高危组患者倾向于表达危险基因,而低危组患者倾向于表达保护基因。

图3 自噬相关的危险因素与肺腺癌患者预后的关系
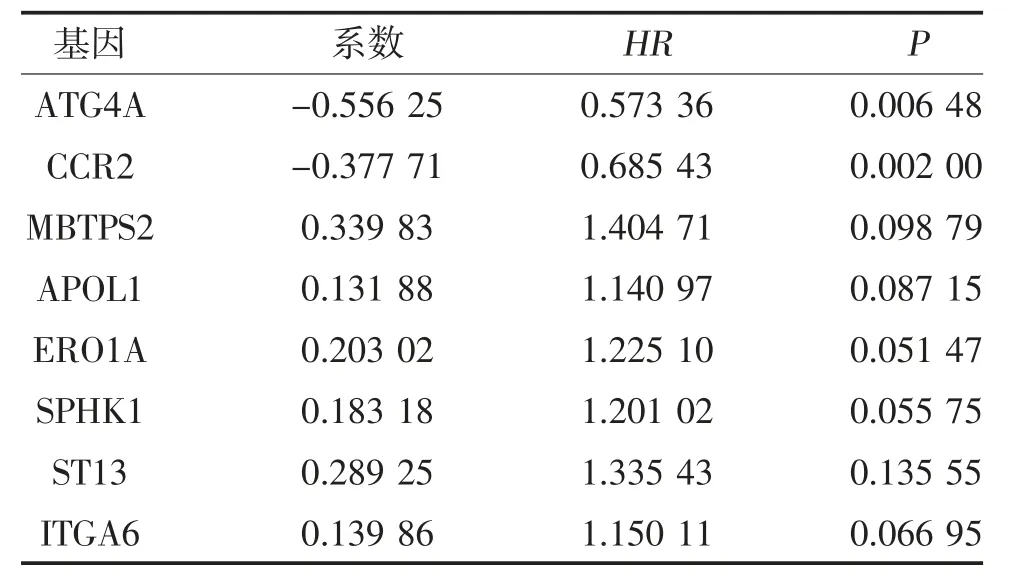
表1 肺腺癌预后基因中的8 个关键ATGs 列表
2.4 自噬在肺腺癌是一个独立的预后指标 单因素和多因素预后分析显示,风险评分是肺腺癌TCGA的独立预后指标(P<0.05),见图4A、4B。Kaplan-Meier 曲线分析显示,高危评分患者的生存期短于低危评分患者,见图4C。与年龄(AUC=0.567)、性别(AUC=0.617)、分期(AUC=0.708)、T 分期(AUC=0.664)、M 分期(AUC=0.506)和N 分期(AUC=0.632)因素相比,风险评分(AUC=0.763)具有更好的预测性能,见图4D。
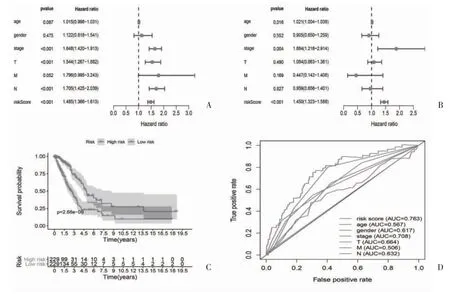
图4 ATGs 与肺腺癌患者的关系
3 讨论
自噬是由一系列ATGs 介导的涉及自噬小体、溶酶体的形成以及细胞器或细胞质的降解过程[7,8]。它可以由缺氧、饥饿、辐射、生长因子信号抑制剂、化疗和靶向药物诱导[9]。近年来,ATGs 在调节细胞内转运、内吞、胞吐、巨噬细胞吞噬和外泌体产生等方面的作用受到越来越多的关注[10-12]。研究表明[13,14],自噬在肿瘤的发生发展中起着双向作用。自噬不仅能有效地抑制肿瘤的凋亡、坏死和炎症进展,而且在抑制肿瘤的发生和发展方面也起到了一定的作用。在这种情况下,自噬是对身体的一种保护作用。肿瘤细胞可以通过自噬途径逃避外源抑制物的杀伤作用,从而使肿瘤继续发展,而在这种情况下,自噬起到了促进肿瘤生长的作用。
本研究分析了从肺腺癌TCGA 数据库中获得的200 个ATGs 的表达情况,同时通过GO 和KEGG 分析研究了分子和生物学途径的富集,GO 富集发现细胞成分和生物学过程与自噬密切相关。从分子功能的角度看,蛋白磷酸酶结合与自噬密切相关。研究发现[15],通过蛋白磷酸酶2A 抑制脯氨酰寡肽酶可以激活自噬。此外,在KEGG 分析中,最重要的途径被富集在自噬中。基于以上结果,特异性自噬可能是肺腺癌发生发展过程中的一种肿瘤促进剂。此外,单因素Cox 回归分析显示,21 个ATGs 与肺腺癌患者的生存有关。随后,通过多因素Cox 回归分析确定了8 个关键ATGs(ATG4A、CCR2、MBTPS2、APOL1、ERO1A、SPHK1、ST13、ITGA6)。研究发现[16],ATG4A对肺癌的预后有重要影响。同时,有报道称[17],miRNA可以调节ATG4A 的表达参与自噬的调节。CCL2 可以与肿瘤细胞表面的CCR2 结合,通过CCL2/CCR2分子轴促进肿瘤细胞的增殖、侵袭等恶性生物学行为[18]。ERO1A 在非小细胞肺癌中高表达,与肿瘤细胞的增殖和迁移有关[19]。SPHK1 也与肺癌的恶性生物学行为有关[20],参与了一些细胞自噬的调节。ST13在正常粘膜组织和相应的肿瘤组织中均有不同程度的表达[21],miRNA 可以在转录后水平调控ITGA6 的表达。另有研究发现[11],miR-126 和miR-143-3p 分别能抑制ITGA6 的表达,从而抑制非小细胞肺癌的侵袭和转移。本研究通过计算ATGs 的mRNA 表达值和风险系数得到风险评分,并根据风险评分对患者进行分层,结果显示自噬基因可作为肺腺癌的独立预后指标,且ROC 分析显示,与年龄(AUC=0.567)、性别(AUC=0.617)、分期(AUC=0.708)、T 分期(AUC=0.664)、M 分期(AUC=0.506)和N 分期(AUC=0.632)因素相比,风险评分(AUC=0.763)具有更好的预测性能。
综上所述,本研究构建了基于8 个ATGs(ATG4A、CCR2、MBTPS2、APOL1、ERO1A、SPHK1、ST13、ITGA6)的预后特征模型,为ATGs 在肺腺癌临床中的应用奠定了基础。自噬基因和预后模型可能为改善肺腺癌患者的生存和预后提供新的分子生物标志物及肿瘤驱动基因,有助于制定个体化治疗策略。
猜你喜欢
保健医苑(2023年2期)2023-03-15
中国临床医学影像杂志(2022年2期)2022-05-25
中成药(2018年7期)2018-08-04
西南军医(2016年3期)2016-01-23
医学研究杂志(2015年12期)2015-06-10
中国当代医药(2015年17期)2015-03-01
郑州大学学报(医学版)(2015年1期)2015-02-27
西南军医(2014年5期)2014-04-25
