
一种含有咔唑基团的荧光化合物光学性能研究
2023-01-13 03:57:18张帮翠陈艳琴窦思虎苟高章杨艳华李福敏
昆明学院学报 2022年6期
张帮翠,陈艳琴,窦思虎,苟高章,杨艳华*,李福敏,邵 林
(1.昆明学院 化学化工学院,云南 昆明 650214;2.红河学院 化学与资源工程学院,云南 红河 661100;3.大理州食品检验检测院 理化检验中心,云南 大理 671000;4.大理州食品检验检测院 色谱分析中心,云南 大理 671000)
通过特异性识别基团受体与目标分子结合,获得荧光信号变化的化学荧光传感器,是化学学科分子研究的热点方向之一.含有碳氮双键基团、二甲胺基团、吡啶基团、喹诺酮基团和吲哚嗪基团的有机化合物中,N和S原子上具有孤对电子,对氢离子进行特异性识别,可作为pH探针[1-3].而酰胺键上的N和O原子都含有孤对电子,也可作为氢离子识别位点.为了探究含有酰胺键基团化合物作为pH探针的可行性,本文设计了一种具有咔唑基团的荧光化合物,其中咔唑基团具有优异的空穴传输能力和刚性平面结构,在分子结构中通常用作电子供体.咔唑衍生物具备较高的荧光发射强度,可用于合成光学传感材料[4].在咔唑基团的3号位引入苯萘基团,以增加分子的空间位阻,进一步改善发光性能.
1 实验部分
1.1 试剂和仪器
所有有机化合物均购于上海麦克林生化科技有限公司;实验合成用有机试剂均购于南京化学试剂股份有限公司,均为分析纯;测试光谱用试剂购于上海麦克林生化科技有限公司,均为光谱纯;超纯水由实验室普利菲尔超纯水机制备.
采用Bruker Avance II-400型核磁共振仪在 25 ℃ 下测试有机化合物的1H NMR和13C NMR;RY-1G熔点仪测试有机化合物熔点;安捷伦Carry Eclipse型荧光分光光度计测试荧光发射光谱;岛津UV-2450型紫外-可见分光光度计测试紫外-可见光谱;安捷伦Cary 640 FTIR型傅立叶变换红外光谱仪测试红外光谱.
1.2 实验步骤
实验合成路线如图1所示,其中化合物A1根据文献[5]合成,合成过程不再赘述.
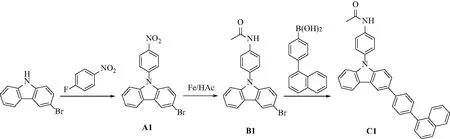
图1 化合物C1的合成路线
1.2.1 化合物B1的合成
N2保护下,在 100 mL 的三口烧瓶中依次加入化合物A1(2.046 g,5.57 mmol)、铁粉(0.924 g,16.5 mmol)和冰醋酸(60 mL),加热至 100 ℃ 搅拌 30 h.冷却至室温,氨水中和至pH为碱性,抽滤,滤液用CH2Cl2萃取(3×100 mL),收集有机层.无水Na2SO4干燥,抽滤,旋干,加入CH2Cl2(70 mL)搅拌,抽滤,滤饼用CH2Cl2冲洗(3×20 mL),旋干得到化合物B1(C20H15BrN2O).熔点:187 ℃.1H NMR(400 MHz,DMSO-d6,δ):10.25(s,1H,-NH-),8.51(d,1H,J=4.0 Hz,ArH),8.31(d,1H,J=8.0 Hz,ArH),7.88(d,2H,J=8.0 Hz,ArH),7.56~7.52(m,2H,ArH),7.48~7.44(m,2H,ArH),7.35~7.28(m,3H,ArH),2.11(s,3H,-CH3).13C NMR(101 MHz,DMSO-d6,δ):169.05,141.19,139.56,139.44,131.32,129.04,127.69,127.51,124.96,123.57,121.97,121.56,120.76,120.71,112.36,112.10,110.32,24.56.FT-IR(KBr压片,cm-1):1 670,1 600,1 544,1 266,571.
1.2.2 化合物C1的合成
N2保护下,在 50 mL 的三口烧瓶中依次加入化合物B1(0.6 g,1.58 mmol)、4-(1-萘基)苯基硼酸(0.397 g,1.6 mmol)、Pd(PPh3)4(0.018 3 g,0.015 8 mmol)、甲苯(30 mL)和蒸馏水(15 mL),加热至 100 ℃ 搅拌 8 h.冷却至室温,甲苯萃取(3×20 mL),收集有机层.无水Na2SO4干燥,抽滤,旋干,柱层层析法提纯(淋洗剂∶V(乙酸乙酯)∶V(石油醚)=1∶2),得到化合物C1(C36H26N2O).熔点:231 ℃.1H NMR(400 MHz,DMSO-d6,δ):10.25(s,1H,-NH-),8.70(d,1H,J=1.6 Hz,ArH),8.39(d,1H,J=8.0 Hz,ArH),8.05(d,1H,J=12.0 Hz,ArH),8.00(t,4H,J=8.0 Hz,ArH),7.92(d,2H,J=8.0 Hz,ArH),7.87(dd,1H,J=2.0 Hz,4.0 Hz,ArH),7.64~7.52(m,8H,ArH),7.49~7.45(m,2H,ArH),7.39(d,1H,J=8.0 Hz,ArH),7.35(q,1H,J=8.0 Hz,ArH),2.14(s,3H,-CH3).13C NMR(101 MHz,DMSO-d6,δ):169.04,141.35,140.47,140.43,139.69,139.29,138.74,133.99,132.34,131.79,131.36,130.83,128.90,128.13,127.65,127.36,127.27,126.95,126.89,126.45,126.13,125.78,125.67,123.81,123.26,121.31,120.77,120.56,119.16,110.61,110.25,24.56.FT-IR(KBr压片,cm-1):1 664,1 600,1 534,1 260.
2 结果与讨论
2.1 核磁与红外表征
从图2(a)可知,化合物B1的-CH3化学位移(δ)为2.11×10-6,-NH-的δ位于10.25×10-6,7×10-6~10×10-6为芳环上氢的化学位移.经偶联反应生成化合物C1后,引入的4-(1-萘基)苯基基团增加了分子中π电子的流动性,产生感应磁场,影响了分子的各向异性效应,-CH3和芳环处于去屏蔽区,δ都向低场移动,其中-CH3的δ为2.14×10-6.
从图2(b)中可以看出,化合物A1中 1 592,1 511 cm-1为-NO2的伸缩振动、561 cm-1为C-Br伸缩振动.形成化合物B1后,-NO2的伸缩振动消失,1 670 cm-1为C=O伸缩振动、1 600 cm-1为N-H面内弯曲振动、1 544 cm-1为N-H弯曲和C-N伸缩振动叠加、1 266 cm-1为C-N伸缩振动和N-H弯曲叠加、571 cm-1为C-Br伸缩振动.经偶联形成的化合物C1中,C-Br伸缩振动消失,1 664 cm-1为C=O伸缩振动、1 600 cm-1为N-H面内弯曲振动、1 534 cm-1为N-H弯曲和C-N伸缩振动叠加、1 260 cm-1为C-N伸缩振动和N-H弯曲叠加.
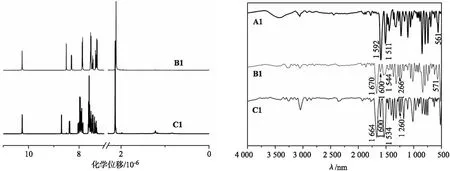
(a)化合物B1和C1的核磁氢谱(DMSO-d6) (b)化合物A1,B1和C1的红外光谱图2 化合物的核磁氢谱和红外光谱
2.2 紫外-可见光谱和荧光发射光谱研究
测试紫外-可见光谱和荧光发射光谱的试剂分别为CH2Cl2,CH3CN和CH3OH等3种极性逐渐增加的溶剂,溶液浓度为1×10-5mol/L,测试结果如表1所示,紫外-可见光谱和荧光发射光谱如图3所示.
从图3(a)中可知,化合物C1在CH2Cl2,CH3CN和CH3OH这3种溶剂中的紫外-可见光谱峰形变化不大,最大吸收波长由分子中S0-S1电子能级下π-π*电子跃迁产生的吸收,如表1所示,摩尔消光系数的数量级在 105L/(mol·cm)-1,说明化合物C1对光的吸收能力较强.与此同时,随着溶剂极性的增加,光谱略微蓝移且吸收强度增加.化合物C1含有N原子和O原子,具有未配位的电子对,存在n-π*态,导致CH3CN和CH3OH等极性分子易于与N原子和O原子上的孤对电子形成分子间氢键,增加了n-π*态跃迁能垒,因此,最大吸收波长发生蓝移.
从图3(b)中可知,化合物C1在CH2Cl2,CH3CN和CH3OH这3种溶剂中的荧光发射光谱峰形变化不大,最大发射波长由π电子从第一单重激发态的最低振动能级经辐射跃迁至基态所致.与此同时,随着溶剂极性的增加,光谱略微红移且发射强度增加.这是由于π电子离域程度随溶剂极性增加而变大,导致激发态与基态间的能带宽度变窄,处于激发态的π电子从第一单重激发态的最低振动能级经辐射跃迁至基态时辐射波长变大,荧光光谱发生红移.此外,化合物C1在溶剂中的Stokes位移较大,说明化合物C1的乙酰胺基团和4-(1-萘基)苯基基团扰乱了分子π-π堆积效应,导致化合物C1可能存在聚集态诱导发射效应.

表1 化合物C1在不同溶剂中的光谱数据
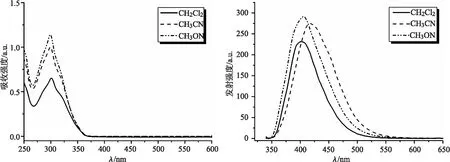
(a)化合物C1在不同极性溶剂中的紫外-可见光谱 (b)化合物C1在不同极性溶剂中的荧光发射光谱 (激发波长为320 nm)图3 化合物C1在不同溶剂中的紫外-可见光谱和荧光发射光谱
2.3 酸碱性对溶液的光谱影响
由于化合物C1中的N原子和O原子具有未配位的电子对,可与质子结合,因此使用HBF4溶液测试其对酸性溶液的敏感性.此外,化合物C1中活泼的酰胺键含有-NH-基团,可使用氨水溶液测试其对碱性溶液的敏感性.测试时化合物C1溶于CH3CN溶液,浓度为1×10-5mol/L,HBF4水溶液和氨水溶液的浓度都为1×10-2mol/L(pH分别为2和12).测试的紫外-可见光谱结果见图4.
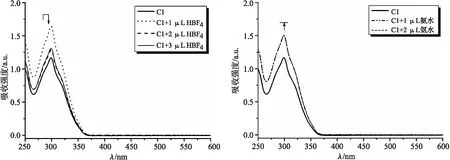
(a)化合物C1对酸敏感的紫外-可见光谱 (b)化合物C1对碱敏感的紫外-可见光谱图4 化合物C1对酸、碱敏感的紫外-可见光谱
从图4(a)可知,随着HBF4的加入,吸收强度呈现先增强后减弱的变化,当加入的HBF4体积为 3 μL 时,吸收强度不再发生变化,且不与化合物C1的光谱重叠.然而,向化合物C1的CH3CN溶液中加入 1 μL 氨水溶液后,吸收强度增大,之后便不再发生改变,如图4(b)所示.同时,无论是加酸还是加碱,化合物C1的吸收峰形无明显变化.以上结果说明,化合物C1对酸更敏感.
荧光发射光谱结果如图5所示.从图5(a)可知,随着HBF4的加入,发射强度并无明显变化,且与化合物C1的光谱重叠.然而,向化合物C1的CH3CN溶液中加入 1 μL 氨水溶液后,发射强度增大,这是由碱性溶液对化合物C1产生光诱导电荷转移所致,随着氨水溶液的增加,发射强度不再发生改变,如图5(b)所示.同时,无论是加酸还是加碱,化合物C1的吸收峰形无明显变化.
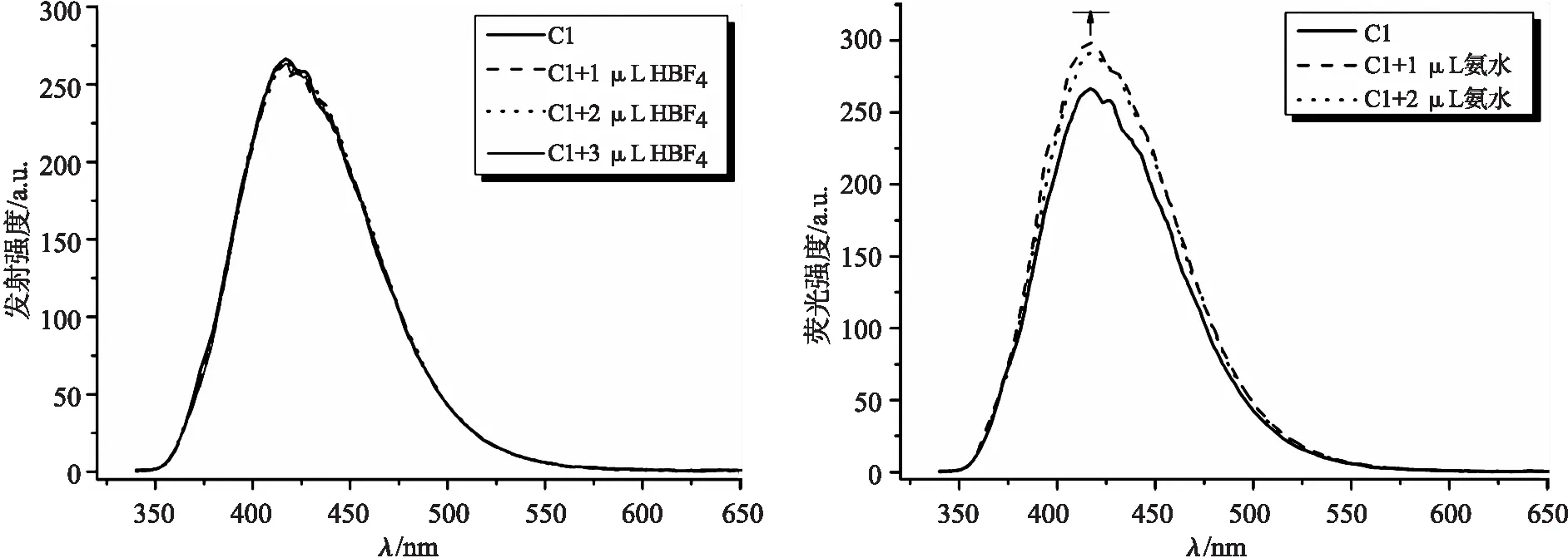
(a)化合物C1对酸敏感的荧光发射光谱 (b)化合物C1对碱敏感的荧光发射光谱图5 化合物C1对酸、碱敏感的荧光发射光谱
2.4 聚集态诱导发光性能
为探究化合物C1是否具有聚集态诱导发射性能,采用CH3CN/H2O混合溶剂控制化合物C1在溶剂中的溶解和聚集,浓度为1×10-5mol/L,实验结果如图6所示.由于水是不良性溶剂,当混合溶剂中含水量从0%(纯CH3CN溶剂)逐渐增加至70%时,处于激发态的激子通过非辐射跃迁返回基态的途径衰减,分子内旋转受限,形成聚集体,化合物C1的荧光发射强度先降低再增大,出现聚集态诱导发射现象[6],且发射波长发生红移,说明形成了分子内电荷转移态.当混合溶剂中含水量从70%逐渐增加至100%时,荧光发射强度下降且红移,说明含水量继续增加后,影响了化合物C1的溶解性,在混合溶剂中出现沉淀,降低了化合物C1的浓度而导致的[7].
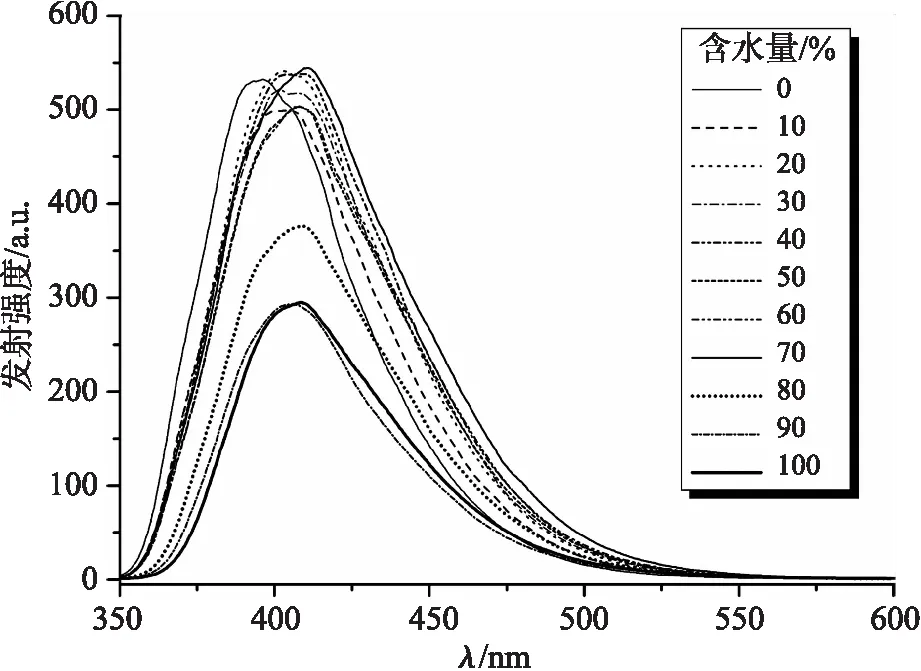
图6 化合物C1在CH3CN/H2O混合溶剂中不同含水量下的荧光发射光谱
2.5 量子化学计算结果
为了更好地解释化合物C1的光学特性,采用Gaussian 09程序,PCM-B3LYP/6-31+G*(在二氯甲烷中)杂化泛函方法,对化合物C1基态时的前线分子轨道(HOMO:最高占据分子轨道,LUMO:最低未占据分子轨道)以及能量、最优构象和静电势表面进行计算,结果如图7所示.
从图7(a)可以得出,化合物C1的HOMO的电荷云密度主要分布于整个分子骨架上,易产生扭曲的分子内电荷转移(TICT),这与聚集态诱发射测试结果中,光谱发生红移实验结果一致.且HOMO-2的电荷云密度与LUMO+1最大重叠,说明位于HOMO,HOMO-1和LUMO-2的电子可被激发跃迁至LUMO和LUMO+1轨道,且化合物C1的最大吸收波长(λmax=301 nm)来源于HOMO-2至LUMO+1轨道的电子跃迁.从图7(b)可以得出,负静电势表面(颜色较深区域)主要位于电负性较大的氧原子部分,说明C=O为化合物C1的吸电子基团,而LUMO的电荷云密度并没有位于此部分,说明化合物C1的咔唑基团(供电子基团)和苯萘部分(共轭基团)的电荷特性比碳基的电荷特性明显,且在LUMO+1和LUMO+2轨道时才出现明显离域.从图7(c)可以得出,咔唑基团与含有酰胺键的苯环和苯萘基团间的二面角分别为56.70°和38.05°,苯萘基团中苯环与萘环间的二面角为56.32°,说明咔唑基团与苯萘基团和酰胺键部分不在同一平面,一定程度上限制了π电子在整个分子平面的流动性,易产生扭曲的分子构象,扰乱分子π-π堆积效应,产生较大的Stokes位移,这与化合物C1的溶剂效应测试结果一致.
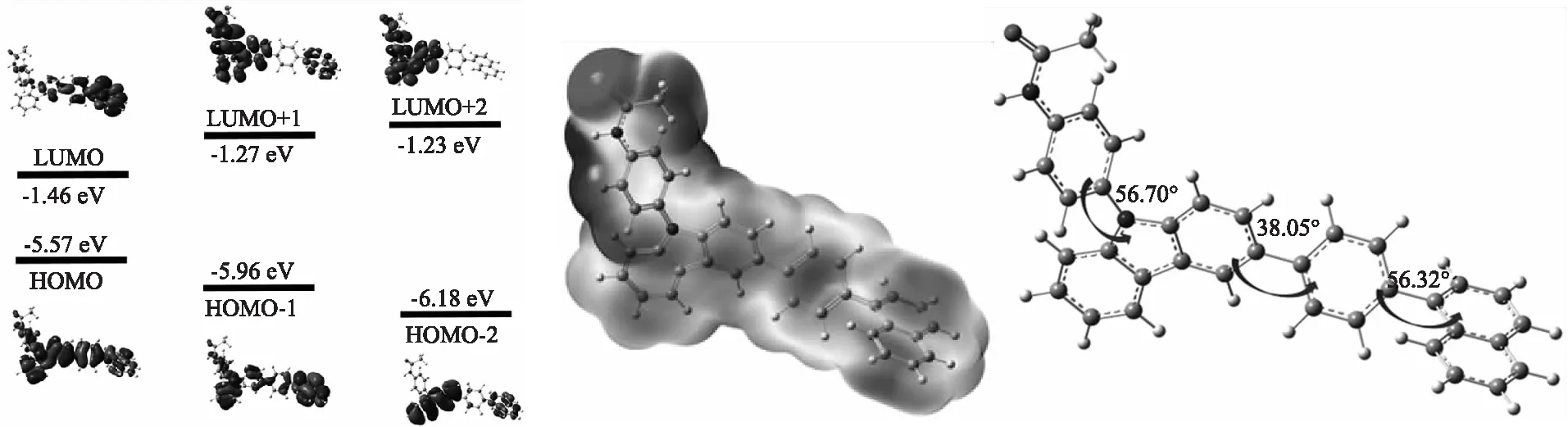
(a)化合物C1的前线分子轨道及能量 (b)化合物C1的静电势表面 (c)化合物C1的最优分子构象图7 化合物C1基态时量子化学计算结果
3 结论
本文合成了一种含酰胺键的咔唑衍生物,并通过核磁氢谱、碳谱和红外测试进行表征,进一步研究其溶剂效应,探究其作为pH探针的可能性.研究结果表明,该化合物具有一定的酸碱敏感度和聚集态诱导发射特性,但仍需进一步进行分子修饰,以接近实际使用效果.此外,本研究可为咔唑有机小分子在离子识别方面的研究提供参考.
猜你喜欢
特产研究(2024年1期)2024-03-12 05:40:56
辽宁化工(2022年8期)2022-08-27 06:03:04
云南化工(2021年11期)2022-01-12 06:06:18
石油炼制与化工(2021年12期)2021-12-14 06:26:38
铜仁学院学报(2018年6期)2018-07-05 09:47:34
塑料助剂(2018年6期)2018-03-25 05:59:16
山东化工(2018年1期)2018-03-10 02:56:49
衡阳师范学院学报(2016年3期)2016-07-10 07:16:27
分析测试学报(2015年9期)2015-12-17 16:44:27
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01 02:53:54
