
Fstl1在肺部疾病中的研究进展
2023-01-11 13:17康海艺刘辉周源
临床肺科杂志 2023年1期
康海艺 刘辉 周源
Fstl1是一种胞外糖蛋白,也是一种分泌型骨形态发生蛋白(BMP)拮抗剂,在文献中,Fstl1有许多不同的名称,如转化生长因子β1诱导蛋白 36(TSC-36)或者卵泡抑素相关蛋白(Follistatin-Related Protein FRP)等。最早是由Shibanuma等[1]利用转化生长因子β1 刺激小鼠成骨细胞系MC3T3-EL而分离出来的一种相对分子质量约35kDa的分泌性糖蛋白,属于BM40/SPARC/Osteonectin家族蛋白的一员,位于3q13染色体,编码308个氨基酸,其氨基末端由12个氨基酸残基组成。该蛋白具有4个N-糖基化位点和2个O-糖基化位点。Fstl1包含一个卵泡抑素(FS)样结构域和一个细胞外钙(EC)结构域,而细胞外钙结构域后紧密相连的C端存在一个与血友因子同源性的结构域。有研究证实,由于Fstl1的EC结构域缺乏钙结合特性,因此Fstl1与SPARC蛋白家族的其他成员在功能上有所不同[1-3]。
一、Fstl1的生物学功能
Fstl1的生物学功能及作用机制目前仍不明确,许多文献已证实Fstl1参与调控细胞发育、增殖、分化、迁移、凋亡、癌变等多种生物学过程。在生长发育方面,Fstl1的缺失可导致小鼠出生后因呼吸衰竭而死亡,或出生后患有多种骨骼相关疾病[4]。Fstl1的表达下调,会导致前体脂肪细胞向脂肪细胞转化,从而导致非胰岛素依赖型糖尿病、高血压、心肌梗塞和某些类型癌症等疾病发生[5]。在肿瘤方面,Fstl1 在不同的恶性肿瘤中的表达是不同的。在鼻咽癌[6]、乳腺癌[7]、肺癌[8]及卵巢癌[9]等肿瘤中Fstl1的表达上调具有抑制肿瘤细胞增殖、侵袭的作用,而在前列腺癌[10]、胶质细胞瘤[11-12]、直肠癌[13]和转移性骨肿瘤[14]等肿瘤中,Fstl1的上调反而会促进肿瘤细胞的增殖、侵袭和转移。
二、Fstl1对肺部发育作用
Fstl1在肺生长发育过程中起着至关重要的作用,已有多个研究证实Fstl1的缺失会导致肺发育不全。Adams等[15]通过对小鼠肺胚胎的观察发现气道周围的肺间充质中有Fstl1的高水平的表达,通过对相邻切片的α-SMA染色及CD31染色发现这种高水平的表达主要存在于气道周围的平滑肌层以及血管壁中。Geng等[16]通过研究Fstl1基因缺陷的小鼠,所有纯合的小鼠在出生后不久死亡。从气管切片上观察到Fstl1-/-缺失的小鼠气管畸形,但这不是Fstl1-/-小鼠呼吸衰竭的主要原因。他们观察到Fstl1-/-的小鼠肺泡球囊上的AEC-Ⅱ细胞是未分化成熟的,其表面活性物质产生减少,从而导致Fst11-/-小鼠肺不张。其机制可能为Fstl1通过负调控BMP4/Smad1/5/8信号调节肺AEC-Ⅱ细胞分化。Sylva等[17]的实验数据与Geng等[16]的实验数据部分相同,进一步证实Fstl1在肺发育中的重要作用。他们通过体外实验发现,加入BMP抑制剂Noggin可以改善Fstl1-/-小鼠肺不张,机制可能是Fstl1的缺失破坏了BMP和FGF信号转导的平衡。因为Fstl1在肺发育中起着拮抗BMP4信号的重要作用,Li等[4]建立了SFTPC-Fstl1转基因小鼠,该鼠在肺发育的所有阶段都表现出明显的 Fstl1 上皮过表达。然而这种过表达并没有改变Fstl-/-小鼠的肺不张。其原因为Fstl1的过表达不能降低小鼠肺中的BMP4诱导的磷酸化Smad1/5/8的水平。随后在成年转基因SFTPC-Fstl1小鼠的肺泡灌洗液中检测到大量Fstl1,表明转基因小鼠BMP信号的无效抑制与上皮细胞顶端分泌Fstl1有关。通过对上皮细胞顶端分泌Fstl1研究,发现只有在上皮细胞的基底外侧同时给予 Fstl1 和 BMP4,才能抑制 BMP4 诱导的 Smad1/5/8 磷酸化。由于纯合子Fstl1基因敲除的小鼠出生时就会死于呼吸衰竭,Tania等[18]通过研究内皮特异性敲除小鼠 (Fstl1-eKO mice)发现70%的Fstl1-eKO的小鼠出生后3周死亡。为了研究其死亡原因,实验研究发现内皮细胞中Fstl1的缺失,阻止了肺小血管中肌动蛋白的合成,从而延缓了肺血管的成熟。其机制为:低水平的Fstl1表达导致BMP介导的Smad磷酸化增加,导致下游靶标Jagged1、endoglin、Gata2和Endothelin-1的升高,从而延缓肺血管成熟。Liu等[19]通过建立Fstl1-LacZ小鼠品系以及Fstl1-/-小鼠品系,发现Fstl1-/-会导致气道平滑肌(ASM)及肺血管平滑肌(VSM)严重畸形,其机制为Fstl1通过关键转录因子SRF和肌钙蛋白正向调节α-SMA的表达和ASM的分化。他们的实验表明Fstl1在ASM的正常形成和VSM的发育中起着重要作用。
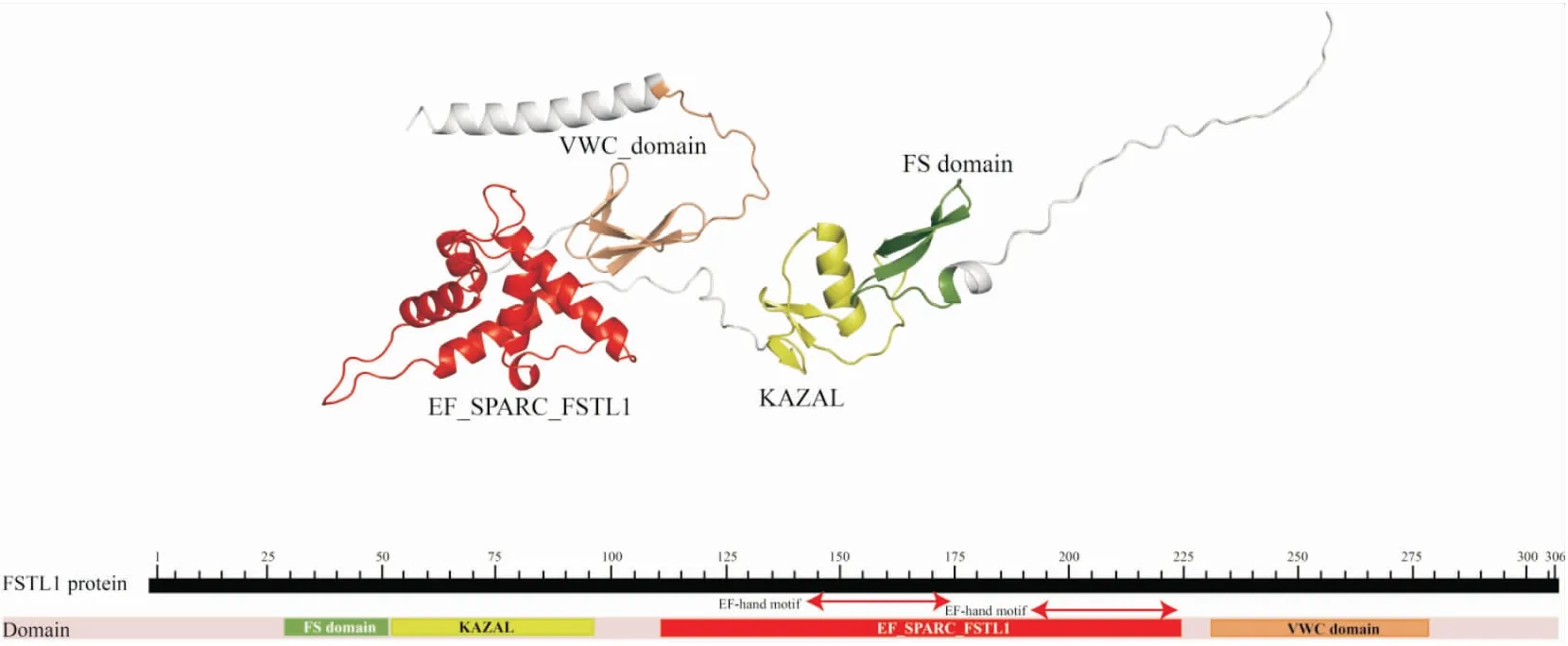
图1 Fstl1蛋白质结构图
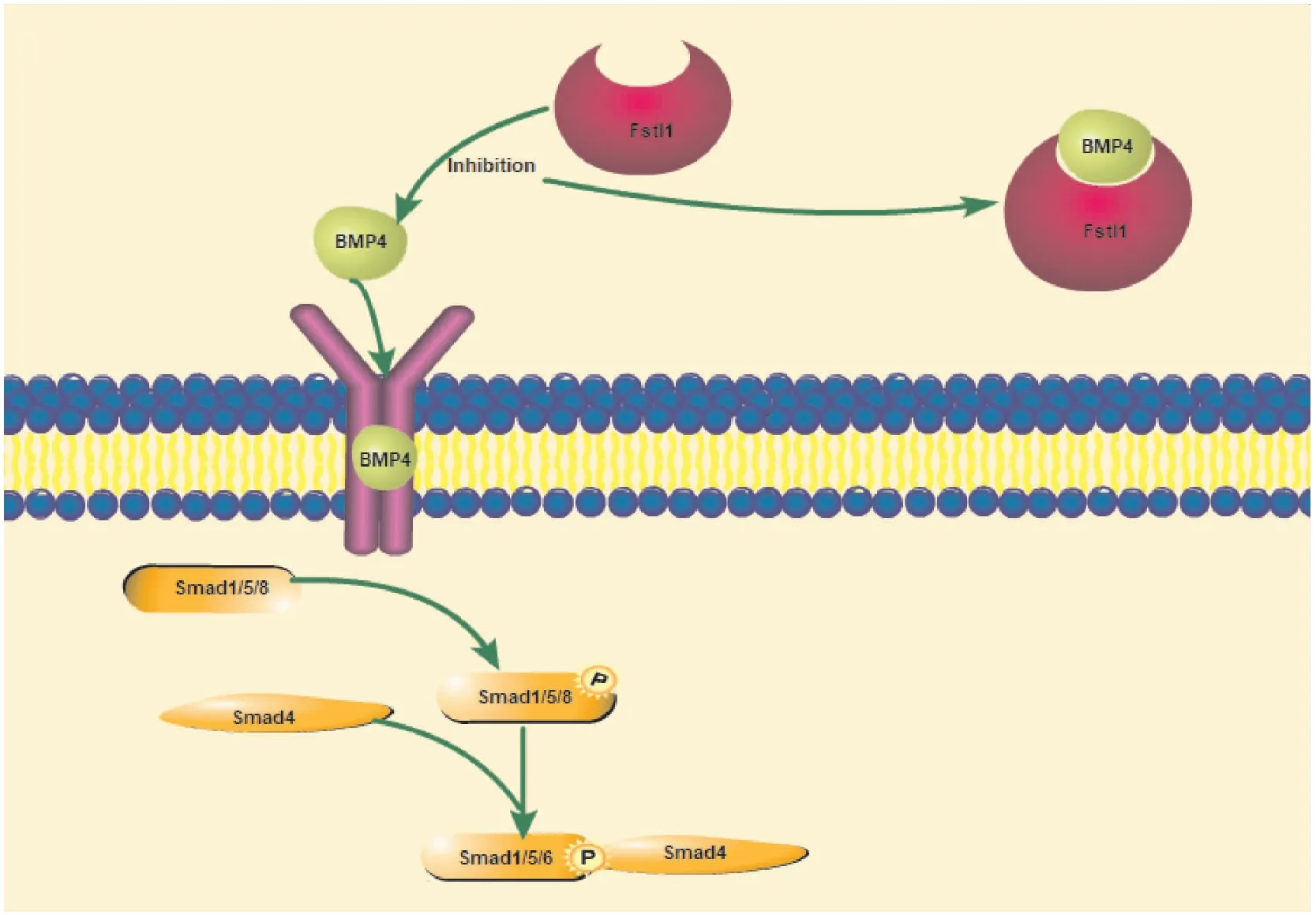
图2 Fstl1调节BMP4信号通路示意图
三、Fstl1对肺部感染性疾病的作用
Henkel等[20]通过对肺炎克雷伯杆菌感染野生型(WT)小鼠和Fstl1亚效等位基因(Hypo)小鼠(一种Fstl1功能降低的小鼠)的研究发现, Fstl1表达降低可以促进IL-17A的表达从而控制肺炎克雷伯杆菌感染,其具体机制不详。Chen[21]等通过体内、外模型研究证实Fstl1表达水平下调可降低骨髓巨噬细胞感染肺炎链球菌后的炎症损伤。其机制为下调Fstl1后可抑制NLRP3和TLR4/NF-κB信号通路,对肺炎链球菌感染所致的炎症损伤具有保护作用。
四、Fstl1对急性肺栓塞(PE)的作用
Liao等[22]对220例急性PE患者进行前瞻性观察队列研究,运用酶联免疫吸附法测定血浆中Fstl1水平,通过多因素Cox回归分析等发现,血浆Fstl1水平与急性PE患者的病情严重程度相关,并可预测30天内死亡的风险。
五、Fstl1对肺动脉高压的作用
Zhang等[23]通过对COPD组、COPD合并肺动脉高压组及对照组患者的血清Fstl1水平进行分析,发现前两组Fstl1水平均高于对照组,且COPD合并肺动脉高压组高于COPD组。在动物实验中,将Fstl1+/-小鼠置于低氧环境,发现Fstl1+/-小鼠右室收缩压、肺动脉肌化和右室肥厚指数增加,随后予Fstl1+/-小鼠尾静脉注射重组人Fstl1蛋白,观察到上述症状得到明显好转。在培养的人肺动脉平滑肌细胞(PASMC)中观察到,Fstl1可抑制缺氧所诱导的PASMCs异常增殖和迁移。为明确其抑制的主要分子机制,通过对缺氧性肺血管重构的关键转导因子:Smad1/5/8、丝裂原激活蛋白激酶(ERK、p38激酶和Jun-N末端激酶)和AMP激活蛋白激酶(AMPK)的磷酸化水平等的实验,发现Fstl1完全敲除后,会导致缺氧的PASMCs中p-ERK的激活,从而加重低氧型肺动脉高压。以上研究证实Fstl1对于低氧型肺动脉高压具有潜在保护作用。Liu等[24]在COPD患者及CS(香烟烟雾)暴露的小鼠模型的血清及肺组织样本的研究发现,Fstl1水平升高且与自噬小体的自噬激活呈正相关,在CS暴露小鼠上使用自噬抑制剂3-MA和/或敲除Fstl1单倍体,发现自噬抑制剂及Fstl1缺失均可减轻CS暴露小鼠的气道炎症和气道重塑反应,具体机制不详,但靶向Fstl1和自噬可能有助于治疗CS诱导的COPD。
六、Fstl1对哮喘疾病的作用
Miller等[25]通过实验发现,Fstl1在重度慢性哮喘的患者肺组织中高水平表达,在哮喘的小鼠动物模型上,M2巨噬细胞、成纤维细胞和上皮细胞可高表达Fstl1,且Fstl1还通过自分泌或旁分泌途径诱导巨噬细胞、成纤维细胞或上皮细胞进一步表达Fstl1。通过体外实验证实Fstl1能直接诱导骨髓中的巨噬细胞表达抑癌素M(OSM)。其机制为Fstl1通过诱导其通路下游OSM来加重慢性哮喘小鼠的气道重塑、嗜酸性粒细胞性炎症和AHR。Liu等[26]在对32例哮喘患者和25例对照组患者的血浆、肺泡灌洗液的Fstl1水平及支气管活检组织的免疫组化染色和定量分析中表明,Fstl1可作为哮喘的血液生物标志物,在哮喘的诊断中有一定的实用价值,并且通过治疗组和未治疗组的血浆Fstl1浓度对比,发现随着哮喘好转,血浆Fstl1水平逐渐降低,表明Fstl1可能促进哮喘患者的气道重构。他们的另一项研究运用不同浓度的Fstl1重组蛋白处理16HBE细胞(人支气管上皮细胞),观察到Fstl1可同时诱导自噬和上皮-间充质转化,在16HBE细胞中使用自噬抑制剂LY294002抑制自噬,发现抑制自噬过程可以减缓Fstl1诱导的EMT和气道重构的进展,并通过动物模型实验进一步证实了此结论;同时他们还发现Fstl1的缺失和LY-294002的治疗对乙酰甲胆碱诱导的AHR具有保护作用[27]。Deng等[28]通过对细胞层面的实验发现,Fstl1的敲除可直接抑制ASM细胞的血小板源性生长因子亚基B(PDGF-BB),从而治疗儿童哮喘。Fstl1的敲除可抑制PDGF-BB诱导的p-EPK和p-AKT蛋白表达水平,减少ASM细胞的增殖和迁移。Wang等[29]通过对哮喘小鼠的研究发现,Fstl1会激活巨噬细胞所诱导NLRP3/IL-1β信号,从而加重过敏性气道炎症反应,而运用siFstl1或MCC950(NLRP3特异性拮抗剂)预处理后,可明显抑制NLRP3和IL-1β的表达,进一步减轻支气管炎症损伤。以上实验研究表明Fstl1有望成为评估治疗哮喘疾病的血液生物标志物,或成为缓解哮喘疾病的靶向治疗药。
七、Fstl1对肺气肿疾病的作用
Henkel[30]等通过研究Fstl1 Hypo小鼠(Fstl1表达降低小鼠)发现Fstl1表达降低会导致小鼠出现自发性肺气肿,且吸烟暴露不会加剧Fstl1 Hypo小鼠的肺气肿情况。为明确机制,作者通过实验,发现Fstl1的表达降低使得肺免疫细胞中的炎症抑制因子Nr4a1/Nur77表达降低,从而直接影响巨噬细胞中的NF-B信号转导。而给予外源性的Fstl1后,会显著增加Nr4a1的表达。该文为深入了解Fstl1信号的候选分子提供了实验依据,为Fstl1在肺部疾病中的治疗潜力提供了实验基础。
八、Fstl1对肺纤维化疾病的作用
研究发现,TGF-β1和BMP信号通路都在肺纤维化的发生和发展中发挥重要作用[31-32]。Dong等[33]通过博莱霉素诱导Fstl1+/-小鼠产生肺纤维化,发现在肺损伤后,Fstl1作为促纤维化因子参与并驱动肺纤维化的发生。而Fstl1的缺失不仅减轻了BLM所致的纤维化,还恢复了TGF-β1/BMP的平衡,从而保护了上皮细胞。随后他们选择Fstl1的中和抗体22B6 mAb进行进一步的研究。结果表明,22B6 mAb可抑制Fstl1,从而维持TGF-β1/BMP的平衡,并调节上皮-间充质转化。Zheng等[34]通过对小鼠正常肺成纤维细胞和博莱霉素处理后14d的小鼠肺成纤维细胞进行检查,发现在BLM处理后的小鼠肺成纤维细胞Fstl1表达更高。通过生物信息分析及体内外实验等发现,在Fstl1启动子-591到-350bp区域内,存在C-jun、SPl结合位点,通过功能丧失和功能获得实验,证实C-jun表达升高,会显著刺激Fstl1 mRNA及蛋白的表达,因此TGF-β1通过诱导Smad3/C-jun通路促进Fstl1表达,从而促进肺纤维化的发生。Chen等[35]对有症状的RIPF(放射性肺纤维化)患者的血清、辐射损伤的恒河猴和小鼠的肺组织中Fstl1蛋白和信使RNA水平进行评估,发现Fstl1表达水平均升高,同时在Fstl1+/-小鼠中同样予以相同剂量的肺辐射,并检测Fstl1的表达水平,与野生型小鼠对照组对比,发现Fst11缺陷对小鼠放射性肺损伤具有保护作用。从野生型小鼠和Fstl1+/-小鼠中分离出原代肺成纤维细胞,用单剂量8 Gy治疗24 h后,通过Western blot检测Fstl1蛋白表达水平表明,Fstl1的缺乏不仅限制了辐射诱导的肌成纤维细胞的堆积,还降低了辐射相关的α-SMA蛋白的水平,从而抑制肌成纤维细胞的分化及细胞外基质的产生。Li等[36]发现单克隆中和抗体(NAB)能够阻断Fstl1所致的肺纤维化。他们通过给博莱霉素诱导的肺纤维化小鼠模型注射两种Fstl1中和抗体:2K6或4D22,发现Fstl1 nAb治疗显著降低了肺纤维化,但Fstl1 nAb不能起到预防肺纤维化的作用。其机制为这两种nAbs阻断了Fstl1介导的间质细胞激活,逆转了肺纤维化和炎症的反应。Jin等[37]发现Fstl1是一种促纤维化蛋白,可通过p38-JNK- Smad2/3信号通路调控TGF - β1促进成纤维细胞的分化、增殖、迁移和侵袭。Fang等[38]在矽肺小鼠模型及矽肺患者血清中测得Fstl1表达上调,通过对使用中和抗体阻断Fstl1的矽肺小鼠模型及Fstl1单倍体缺失的矽肺小鼠模型相关实验数据对比,发现都可减轻小鼠体内二氧化硅诱导的肺部炎症及纤维化。阻断或敲除Fstl1可降低体内NLRP3的表达、caspase-1活性和随后的IL-1β分泌。这些研究表明,Fstl1有潜力作为肺纤维化的治疗工具。
九、Fstl1与非小细胞肺癌的关系
目前关于Fstl1与非小细胞肺癌关系的研究较少,对Fstl1在非小细胞肺癌组织中的表达情况、作用机制等,各研究结果也不一致。有研究表明,连接蛋白基因已经被归类为肿瘤抑制基因[39-40]。Zhao等[41]发现,Cx43除了具有抑制肿瘤生长的作用外,还具有抑制肿瘤侵袭和转移的作用,而Cx43的抑制作用是通过调节Fstl1的分泌而抑制PG细胞的侵袭和转移,Cx43的抑制作用依赖Fstl1的分泌。Ni等[42]检测了一组非小细胞肺癌细胞系和肺正常上皮细胞系中Fstl1的表达,发现Fstl1在NSCLC细胞中的表达较正常对照组下调。Fstl1改变了Fas/FASL、caspases和MMPs等与细胞凋亡和侵袭相关的关键因子,从而通过改变细胞周期和增加凋亡来抑制NSCLC细胞的增殖。
也有研究发现,Fstl1的下调强烈地预示着肺腺癌患者的预后较差。Chiou等[8]通过实验发现细胞外抗体中和Fstl1可增加肺癌细胞的迁移/侵袭能力,而加入重组Fstl1蛋白则可降低肺癌细胞的体内外转移能力。运用Fstl1治疗可以有效地阻止肺癌细胞的转移进展。其机制为Fstl1直接与新生SPP1结合,抑制其被基质金属蛋白酶-3/7或凝血酶蛋白酶蛋白水解成活性SPP1的过程,阻止SPP1诱导的转移癌细胞中avb3整合素和CD44的活化,从而抑制了LUAD(肺腺癌)细胞的转移进程。
而在有吸烟史的的LUAD患者中,Fstl1的低表达更为常见。 Chiou等[43]发现Fstl1通过调节Fstl1-BMP4-Smad通路在肺腺癌中发挥重要作用。而低表达的Fstl1、BMP4及Smad在肺腺癌中显示出预后不良的趋势,在有吸烟史的LUAD患者中更常观察到低Fstl1,BMP4和Smad4表达。通过用尼古丁处理正常细胞BEAS2B和肺癌细胞株BEAS2B发现尼古丁能通过ERK/MAPK信号通路促进肺癌细胞的增殖。而Fstl1可抑制尼古丁诱导的肺癌细胞增殖。
相反的,有研究发现,阻断Fstl1可以有效诱导抗肿瘤免疫,特别是在转移性肿瘤中。Chie等[44]运用转移模型比较了抗Fstl1单克隆抗体和IC抑制剂(抗CTLA4、抗PD1和抗PDL-1单克隆抗体)的抗肿瘤效果,在诱导抗转移免疫方面,抗Fstl1治疗优于IC抑制剂治疗,且阻断Fstl1能更有效抑制老年小鼠的癌症进展。在肿瘤培养中加入抗Fstl1单抗也可抑制细胞增殖并增强对CTL杀伤的敏感性。实验结果提示Fstl1和DIP2A共阳性的肿瘤细胞可以通过自分泌Fstl1的方式来维持肿瘤恶性特性,而且还可以损害宿主的免疫功能。因此,破坏Fstl1-DIP2A轴可以通过控制肿瘤特性和免疫平衡来有效地消除癌症。抗Fstl1单抗可用于癌症的治疗,其机制不同于临床上以IC通路为靶点的机制,至少在非小细胞肺癌中是如此。
也有研究表明,阻断Fstl1可诱导非小细胞肺癌(NSCLC)细胞死亡。Bae等[45]在NSCLC细胞(NCI-H460、NCI-H2228和A549)中根据Western blotting测定的PARP裂解结果,发现细胞死亡率最高的为NCI-H460细胞,随后用NCI-H460细胞来评估Fstl1基因敲除诱导的细胞死亡发现,Fstl1基因敲除可诱导细胞周期阻滞于G2/M期,细胞周期蛋白CDK表达上调。进一步研究了细胞周期进程的影响,发现阻断Fstl1抑制了ERK1/2的激活,从而抑制了Bim磷酸化导致Bim亚型的积聚。
以上研究表明 Fstl1 对非小细胞肺癌具有双调控作用,既可抑制癌细胞增殖、侵袭和迁移的作用,也可通过敲除Fstl1从而有效诱导抗肿瘤免疫或诱导癌细胞的死亡,但具体机制仍需进一步研究。
十、结束与展望
目前,Fstl1在肿瘤及炎症性疾病中具有大量的研究,但对于其与肺部疾病的关系及其作用机制的研究仍较少且具有争议。而明确 Fstl1与肺部疾病的关系、作用和调控机制,可能会为肺部疾病的治疗、诊断及预后提供理论依据。尤其是在肺纤维化疾病哮喘及非小细胞肺癌中,仍需要进一步研究 Fstl1的具体作用机制及调控通路。这将会为以后诊断或治疗肺纤维化、哮喘及非小细胞肺癌提供一个新方向。
猜你喜欢
中老年保健(2022年2期)2022-11-25
保健医苑(2022年1期)2022-08-30
中国临床医学影像杂志(2022年2期)2022-05-25
昆明医科大学学报(2022年4期)2022-05-23
中国药学药品知识仓库(2022年1期)2022-03-23
中老年保健(2021年6期)2021-08-24
昆明医科大学学报(2021年4期)2021-07-23
医学研究杂志(2015年12期)2015-06-10
天津医科大学学报(2015年3期)2015-06-05
郑州大学学报(医学版)(2015年1期)2015-02-27
