
四种全基因组测序用文库构建方法的效果比较
2022-12-30 09:33:54杨漫漫沈俊然
湖北农业科学 2022年21期
杨漫漫,陈 涛,沈俊然,王 然,李 勇
(深圳市华大农业应用研究院∕深圳市动物基因组辅助育种工程实验室,广东 深圳 518083)
低覆盖度重测序近年来在群体遗传学分析[1,2]、基因组选择[3,4]等方面的应用越来越广泛。文库质量对数据质量和分析效果有很大影响[5],覆盖度低时尤为明显,而片段化是文库制备的第一个关键步骤。目前,片段化技术主要为物理打断法和酶切打断法,超声是物理打断法中最为常用的方法,而酶切打断法主要分常规片段化酶和TN5转座酶[6]。此外,为了解决文库制备中由于PCR扩增导致的bias和copy errors等[7,8],无需PCR的PCR-free建库方式也被广泛应用[9,10]。长期以来,测序文库的构建主要依赖人工操作,但文库构建受到3方面的挑战:流程复杂度、过程污染和单个建库成本。升级建库技术,简化操作流程,减少人与试剂和样本的交互,在降低污染风险的同时,也减少了人工、样本、试剂的投入以及缩短操作时间,进而使得每个样本的投入成本也大大降低,这是测序流程自动化发展的基本趋势[11]。本研究探索了不同建库方式在低覆盖度重测序中的数据表现,并对这些数据提供一个粗略的评估,为自动化建库及测序流程优化提供部分数据参考。
1 材料与方法
1.1 试验材料
1.1.1 供试样本供试材料为大白猪耳组织,来源于温氏清远原种场。
1.1.2 仪器与试剂S1000型Bio-rad PCR仪,美国Bio-rad公司;Qubit3.0型荧光定量仪、Qubit 1X dsDNA HS kit(Q33230),美国Thermo Fisher公司;MSP-960型高通量自动化样本制备系统、MGIseq2000型基因组的测序仪、MGIEasy通用DNA文库制备试剂套装(1000006986)、MGIcare染色体异常检测试剂盒(1000005279)、酶切PCRfree DNA文库制备试剂盒(1000013455)、MGIseq2000RS高通量快速测序试剂套装(1000013155),深圳华大智造科技股份有限公司;DNA磁珠法提取试剂盒(GO-BTCD-100),长春市志昂生物科技有限公司。
TN5原料酶由深圳华大生命科学研究院提供。
1.2 试验方法
耳组织样本使用组织DNA磁珠法提取试剂盒进行DNA提取,琼脂糖凝胶电泳和Qubit dsDNA HS检测试剂盒进行质量控制。
超声打断是文库构建的经典方法,采用MGIEasy通用DNA文库制备试剂套装,初始样本投入量约500 ng。MGIcare染色体异常检测试剂盒操作步骤简单,对DNA总量要求低(50 ng),能够适用自动化设备。酶切PCRfree DNA文库制备试剂盒DNA投入无需均一化,全流程无PCR错误累积,适合全程自动化。转座酶建库用于NGS测序近年来应用广泛,根据Picelli等[12]、Zan等[13]的方法进行接头序列改造后适应DNBseq平台,初始样本投入量在100 ng。详细的文库构建流程见图1。

图1 不同方式文库构建流程
构建好的文库在MGIseq2000测序仪上采用PE 100进行0.5-10x深度测序。
1.3 数据分析
原始数据下机后,过滤掉碱基质量值低于20且比例超过30%的read。从4种建库方式的数据中随机选取6个样品,使用软件seqtk分别抽取15、30 Mb reads用于后续分析比较。
将过滤的clean Data比对到猪参考基因组序列(sus scrofa 11.1),参考基因组信息经处理,去除未定位序列和线粒体序列,有效基因组大小为2.45 Gb。Picard用于标记PCR重复序列后统计比对信息。GATK默认参数用于变异检测,过滤掉假阳性位点后统计二等位SNP位点信息。
使用26个无关群体的大白猪重测序数据(深度12×-15×)的SNP集作为参考panel,使用beagle软件分别对15、30 Mb数据的SNP集填充到全基因组水平,而后将填充的SNP位点和对应10×样本的SNP信息进行比较,过滤掉原始个体SNP的缺失位点,计算相同个体间皮尔逊相关系数。
2 结果与分析
2.1 重测序数据质量统计
为了比较不同建库方法获得数据的一致性,选用经超声打断和不同酶切打断(MGIcare、PCRfree和TN5酶法)获得测序文库,在MGIseq 2000平台上采用PE 100进行高通量测序(表1),分别获得200.00、307.69、125.00 Gb和1.05 Tb数据,数据产量及变异范围符合预期。从表1的Q20和Q30数据可以看出,4种建库方法获得的数据质量较高,没有明显差异,其中Q20>97%,Q30>89%,GC含量为41.34%~44.17%,GC含量TN5组较高,可能与转座酶的偏好性有关[14]。

表1 不同建库方法的测序质量信息
2.2 不同建库方法获得的测序数据比较
对不同建库方法获得的测序数据进行比对(表2),发现比对率和惟一比对率分别能达97%和94%以上,其中PCRfree建库组的惟一比对率最高,达96.56%;其他指标,如错配率、重复率等都在正常范围,相比而言,PCRfree建库组在所有组中的重复率最高。此外,覆盖度随着测序深度的升高而上升,0.5×覆盖度约30%,1×覆盖度为44%~68%,2×以上覆盖度超过80%。
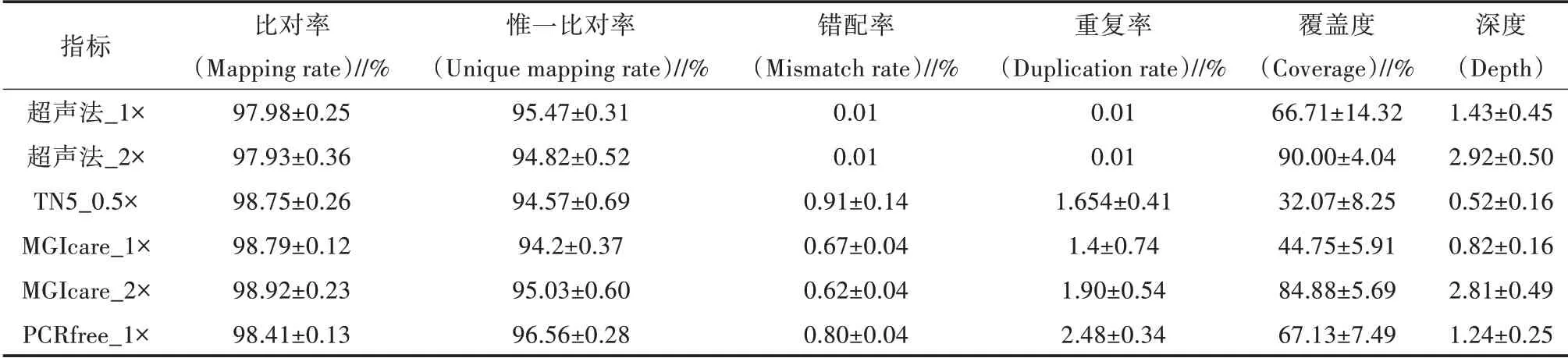
表2 不同建库方法的测序指标分析
将不同建库方法分为2组,一组为手工组(超声法、TN5),一组为自动化组(MGIcare、PCRfree),并对这2组测序数据进行比较分析。从个体数据各项指标的分布上看(图2),自动化建库数据更集中,波动性小于手工建库数据。

图2 手工建库和自动化建库指标分布
2.3 抽成数据分析
为了在同一水平上比较不同建库方式下数据的各项指标,每种建库方式选择6个个体,分别随机抽取15、30 Mb reads(对应测序深度约为0.5×和1×)进行分析(图3)。数据均一化后,除覆盖率和重复率指标外,其他指标在不同建库方法下一致性均较好。覆盖度的波动与建库插入片段的长度以及测序随机性带来的个体间差异有关。从图3可以看出,插入片段越短覆盖度越低,如MGIcare在所有建库方法中插入片段最短,为100~200 bp。均一化后,不同组重复率虽然有波动,但所有组都在正常范围内。

图3 15、30 Mb数据下各项指标分布
利用GATK软件进行变异检测,在0.5×的数据下,不同建库方法获得SNP数量为2 922 270~4 181 825,其中MGIcare获得的SNP数量最高,PCRfree最低(图4a)。在1×的抽成数据下,获得的SNPs数量为6 532 715~7 375 447,不同组SNP数量分布的趋势与0.5×类似。对检出的SNPs进行填充准确性检验,发现利用高深度基因组测序个体进行基因型填充的准确性达74%以上,且0.5×与1×数据填充的准确性相当(图4b)。

图4 4种建库方式变异检测与填充准确性
3 讨论
高质量文库的高效制备在NGS研究中发挥着重要作用,DNA样本片段化是文库制备的第一个关键步骤,包括不同的片段化方法和不同的制备流程。本试验在同一个测序平台上测试了利用不同片段化方法和文库构建方式获得了低覆盖度重测序数据。数据分析结果表明,4种不同片段化方法获得的测序数据在质量和指标上比较一致。尽管研究结果中PCRfree的重复较高,但仍然处于DNBseq平台的正常范围[15]。研究表明,重复不仅与PCR有关,还受到基因组复杂度、碱基组成、连接效率、滚环复制以及光学分辨率等方面影响[6]。MGIcare方法由于其片段化酶的特性导致插入片段的长度偏低(mean length=160 bp),同等数据量条件下覆盖度相对较低,但在变异检测方面更具优势。TN5转座酶的文库制备方法简单、高效,更具有性价比[16]。在测试数据中,TN5酶组操作流程最简单,耗时最少,得到与其他组相似的质量和指标分布数据。此外,还比较了不同建库方式的差异,发现手工建库和自动化建库相比,自动化数据显示出更好的一致性。这与流程中减少了人为操作引起的数据变异有关,对未来大规模推广自动化建库有很好的参考作用。对低深度数据进行填充,与高深度数据相比,低深度填充准确性超过74%。尽管远低于预试验的大规模低深度的数据(99.1%),这可能与高深度参考群与低深度测序群体的亲缘关系、群体规模、LD等有关[17,18]。研究比较了不同的片段化方法和建库方式获得的测序数据,为后期规模化利用低成本的低深度测序技术提供了数据参考。
猜你喜欢
科学技术创新(2022年30期)2022-10-21 14:01:24
农业与技术(2021年23期)2021-12-14 09:03:32
猪业科学(2021年3期)2021-05-21 02:05:36
幽默大师(2020年10期)2020-11-10 09:07:22
原子与分子物理学报(2020年5期)2020-03-17 06:59:18
中华诗词(2019年1期)2019-11-14 23:33:56
猪业科学(2018年4期)2018-05-19 02:04:31
测绘科学与工程(2017年3期)2017-08-16 02:46:00
贵州师范学院学报(2016年1期)2016-12-01 03:53:36
河南科技(2014年5期)2014-02-27 14:08:42
