
超高效液相色谱-串联质谱法同时测定化妆品中50种非法添加药物
2022-12-30 11:52廖文榕黄健浩
分析测试技术与仪器 2022年4期
廖文榕,黄健浩
(福州市食品药品检验所,福建 福州 350007)
近年来,化妆品中非法添加禁用化学药物的现象越发引起国家市场监督管理部门的重视. 广东省药品检验所[1]对2010至2020年期间1 180批次不合格的化妆品进行分析,含非法添加抗感染药物及抗组胺药物的不合格化妆品分别为515批次及46批次,占总不合格项目比率的36.04%及3.22%. 按照《化妆品安全技术规范》(2015年版)[2](以下简称《规范》)中的标准方法,检出16种抗感染及抗组胺药物. 此外,按照非标准方法检出夫西地酸和甲氧苄啶2种抗生素药物. 化妆品中非法添加的禁用药物具有不确定性,不同生产企业可能添加不同的禁用药物. 目前化妆品标准方法所检测的组分无法充分涵盖市场上易添加的抗感染药物组分,且检测方法多样,操作繁琐,检测多组分的成本高昂.
红霉素、莫匹罗星、夫西地酸、多粘菌素B、特比萘芬、甲氧苄啶、伊曲康唑等抗生素药物常作为通用外用软膏剂或口服制剂协同用于皮肤的抗感染治疗. 西咪替丁和雷尼替丁等抗组胺药物(H2受体拮抗药物)具有抗雄性激素、抑制皮脂分泌的特性,可与抗菌药物联合用药治疗痤疮等皮肤病[3]. 这些药物属于处方药,不法厂商可能在宣称具有“祛痘”功效的化妆品中违禁添加这些常见的化学药物,甚至在儿童化妆品中也检出用于抗感染治疗的化学药物. 而在《规范》[2]以及国家药品监督管理局2019年第66号通告附件2中发布的关于《化妆品中抗感染类药物的检测方法》[4]中尚未见到化妆品中上述成分的检测方法.
液相色谱与串联质谱联用(HPLC-MS/MS)是快速发展的一种分析技术,具有检出限低、选择性高、灵敏度高等特点,尤其在组分分离、结构鉴定、定性筛查、定量分析中具有优势,已广泛应用于化妆品中化学药物非法添加筛查领域[5-6]. 近年来,文献所报道的多粘菌素B(多粘菌素B1及多粘菌素B2)[7-9]、夫西地酸[10]、甲氧苄啶[10]、莫匹罗星[11-12]、特比萘芬[13-16]、西咪替丁[3,17]、雷尼替丁[3,17]、红霉素[18-22]、伊曲康唑[23-24]等抗生素及抗组胺药物大都采用HPLC-MS/MS法进行单组分测定或少数几种组分同时测定的方法学研究. 但对化妆品中上述10种组分及甲硝唑等40种组分(化合物名称如表1所列)同时检测的方法未见报道.
本研究在标准文献包含的36种组分基础之上,拓展考察《规范》[2]“2.1 氟康唑等9种组分”中包含的萘替芬、益康唑、咪康唑、环吡酮胺,以及常用的皮肤病治疗药物:红霉素、莫匹罗星、夫西地酸、多粘菌素B(多粘菌素B1及多粘菌素B2)、甲氧苄啶、特比萘芬、伊曲康唑、西咪替丁和雷尼替丁等14种组分,共计50种禁用化学药物(含48种抗感染药物和2种抗组胺药物,化合物名称如表1所列)同时测定的方法学研究.
本研究通过考察常见基质(如:液态水基、乳液和膏霜)化妆品,探索上述50种非法添加化学药物的UPLC-MS/MS定性筛查及定量分析方法. 氟康唑、西咪替丁等50种化学药物采用同一种液相条件进行分离. 同时,简化环吡酮胺的前处理方式,优化多肽类大分子(分子量超过1 000)硫酸多粘菌素B(多粘菌素B1及多粘菌素B2)的质谱条件,考察不同基质对结果数据的影响. 本方法在对现行标准进行整合的基础上,增加了10种常用于皮肤抗感染治疗的化学药物,为化妆品中禁用物质的监督管理提供更为全面、快速的筛查方法,也为化妆品标准方法的改进提供技术参考.
1 试验部分
1. 1 仪器与试剂
超高效液相色谱仪 1 290 UPLC(Agilent公司)串联AB SCIEX QTRAP 5 500三重四极杆复合线性离子阱质谱仪(AB SCIEX公司);Biofuge Stratos高速冷冻离心机(Fisher Scientific公司);XS105DU双量程精密电子天平(梅特勒-托利多公司);VortexGenius3漩涡混合器(德国IKA公司);XM-1000UVF超声波清洗器(小美超声仪器公司);Milli-Q-Reference超纯水机(默克密理博公司);甲醇、乙腈和四氢呋喃(HPLC级,德国Merck公司),甲酸(HPLC级,阿拉丁公司),氯化钠及乙二胺四乙酸二钠(均为GR级,国药集团化学试剂公司).
标准物质:硫酸多粘菌素B(多粘菌素B1及多粘菌素B2的混合物)(德国Dr. Ehrenstofer公司),红霉素(Bepure,北京曼哈格生物科技有限公司),伊曲康唑、莫匹罗星(中国食品药品检定研究院),其余标准物质(FirstStandard®,美国A Chemtek公司),纯度均大于91%.
30批化妆品均为市售品.
1. 2 标准溶液的配制
1. 2. 1 标准储备溶液
准确称取各待测组分标准品约10 mg(精确至0.01 mg),置于10 mL棕色容量瓶中,使用甲醇(其中伊曲康唑加入2 mL四氢呋喃助溶)配制成质量浓度约1 mg/mL的标准储备溶液(−18 ℃保存). 准确移取各标准储备溶液适量,使用甲醇配制成10 µg/mL的混合标准溶液.
1. 2. 2 标准溶液
使用乙腈(0.5%甲酸)-2 mg/L乙二胺四乙酸二钠水溶液(EDTA-2Na),体积比为1∶1,将混合标准溶液配制成质量浓度分别为10、20、40、60、80、100 ng/mL标准工作溶液.
1. 2. 3 基质匹配标准溶液
分别称取6份化妆品基质空白样品0.2 g(精确到0.000 1 g),加入混合标准溶液,按1. 3项下样品的制备同法进行处理,配制成质量浓度分别为10、20、40、60、80、100 ng/mL的基质匹配标准工作溶液.
1. 3 样品的制备
准确称取化妆品样品0.2 g(精确到0.000 1 g),置于15 mL离心管中,加3 mL饱和氯化钠溶液,涡旋分散均匀,加5 mL乙腈(含0.5%甲酸),涡旋30 s,超声提取30 min,涡旋混合30 s,以8 000 r/min转速0 ℃冷冻离心5 min,吸取上清液,加EDTA-2Na水溶液(2 mg/L)定容至10 mL容量瓶,混匀后经0.22 µm滤膜过滤,滤液为待测试样.
1. 4 质谱条件
质谱条件:电喷雾离子源(ESI);离子喷雾电压(IS):5 500~4 500 V;离子源温度(TEMP):500 ℃;多反应监测(MRM)扫描,气帘气(CUR):0.24 MPa,碰撞气(CAD):0.24 MPa,雾化气(GS1):0.34 MPa,辅助气(GS2):0.34 MPa. 去簇电压(DP)及碰撞电压(CE)等质谱参数设定如表1所列.
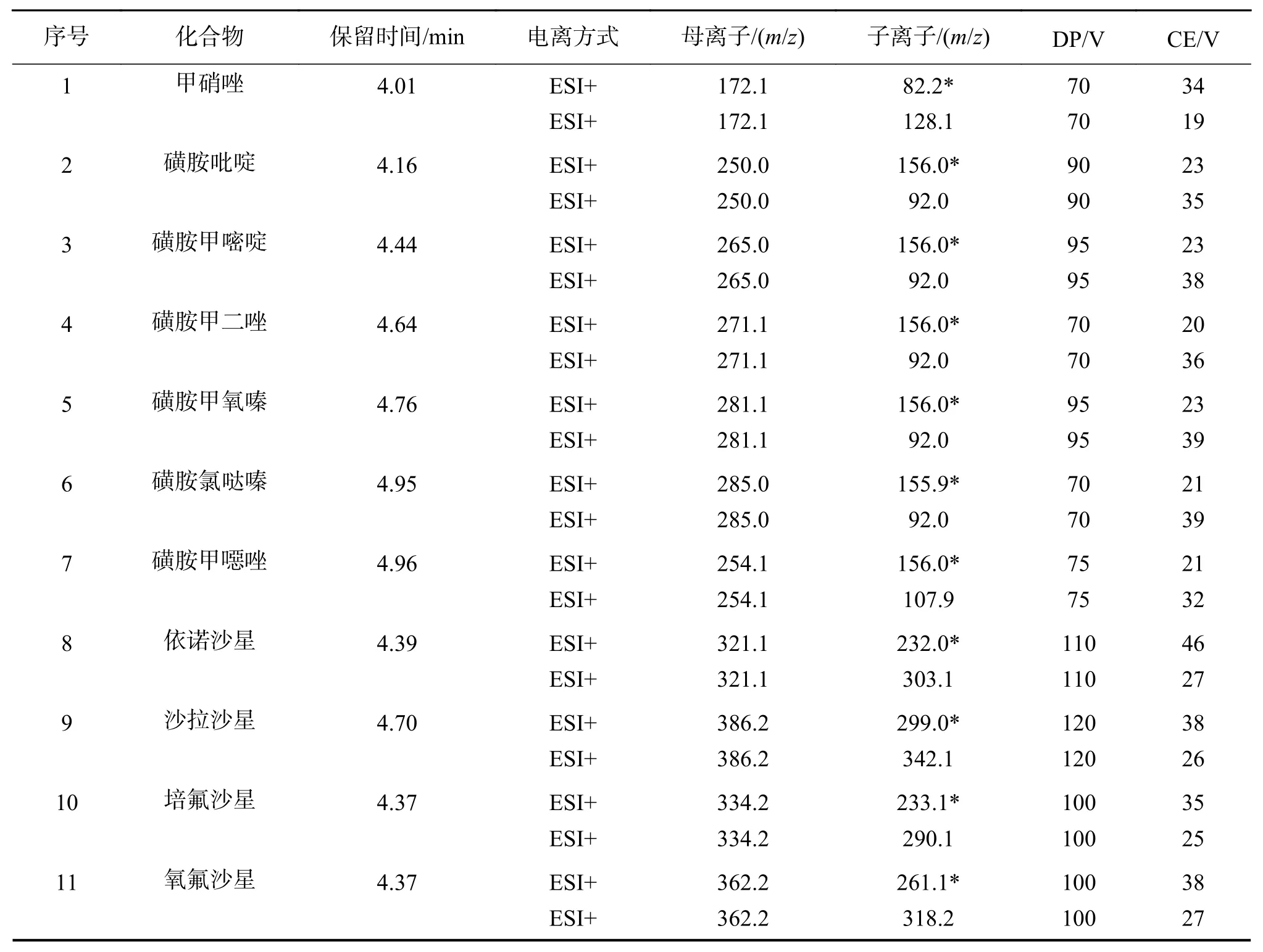
表1 50种化合物的质谱参数Table 1 Mass spectrometry parameters of 50 compounds

续表 1

续表 1
1. 5 色谱条件
CAPCELL CORE C18色谱柱(150 mm×3.0 mm,2.7 µm);流动相A:甲醇,流动相B:0.1%甲酸水溶液,梯度洗脱;柱温:40 ℃;进样量:2 µL,流速:0.3 mL/min. 梯度洗脱程序如表2所列.

表2 流动相梯度洗脱程序Table 2 Gradient elution procedure of mobile phases
2 结果与讨论
2. 1 质谱条件的优化
根据50种化合物的电离性质,分别在ESI+及ESI-离子化模式下,以注射泵连续进样方式,寻找目标化合物的母离子信号,确定定量子离子及定性子离子信息,同时优化DP、CE等质谱参数. 氯霉素和夫西地酸在负离子模式下响应值最大,准分子离子为[M-H]–. 硫酸多粘菌素B是常用的多肽类阳离子型表面活性剂抗生素,分子量超过1 000的为多粘菌素B1和多粘菌素B2的混合物. 带有多电荷,在正离子模式下容易质子化,碎裂后得到的可能是带有两个电荷、三个电荷或者更多电荷的离子. 最终选择响应值大的[M+2H]2+峰(m/z602.7)和[M+3H]3+峰(m/z402.1)作为多粘菌素B1的母离子,以及[M+2H]2+峰(m/z595.7)和[M+3H]3+峰(m/z397.5)作为多粘菌素B2的母离子,并确定相应的子离子. 其余46种化合物均在正离子模式下响应值最大,准分子离子为[M+H]+. 质谱参数信息如表1所列.
2. 2 色谱柱及流动相的选择
影响液相色谱分离的3个主要因素为化合物属性、色谱柱及流动相. 待分析化合物确定后,考察适合的色谱柱及流动相分离条件.
试验考察了色谱柱Agilent ZORBAX SB-Aq C18(100 mm×2.1 mm,1.8 µm)及CAPCELL CORE C18(150 mm×3.0 mm,2.7 µm)对50种化合物的分离效果. 结果表明:目标化合物在CAPCELL CORE色谱柱上的峰型更尖锐、对称,响应值更强,故选择核壳色谱柱CAPCELL CORE C18进行分离检测(50种化合物的总离子流谱图如图1所示).

图1 50种化合物的总离子流图Fig. 1 Total ion chromatogram for 50 compounds
参考标准[4]中的流动相组成:乙腈-5 mmol/L乙酸铵溶液(甲酸调至pH值为4.0),多粘菌素B未见色谱峰出现. 同时考察了不同流动相的组成:乙腈-水(含0.1%甲酸)、乙腈-水(含0.1%三氟乙酸)、乙腈-5 mmol/L乙酸铵溶液(含0.1%三氟乙酸)和甲醇-水(含0.1%甲酸)的分离效果和质谱响应值.由于多粘菌素B含有多个氨基和羧基,易和C18柱中的硅醇基发生相互作用,导致拖尾. 甲酸可以使色谱柱中的硅醇基质子化,减弱其和多粘菌素B之间的相互作用. 同时在正离子检测模式下,甲醇对酸碱性强或电负性强的化合物具有更高的选择性.试验最终采用甲醇-水(含0.1%甲酸)为流动相,并对梯度洗脱程序进行优化,大大提高了多粘菌素B的响应值,且所有正离子及负离子待测化合物均能获得良好的色谱峰型和质谱响应.
2. 3 前处理方法的选择
由于环吡酮胺中的N-OH基团会与分析系统中存在的微量金属离子发生强螯合作用[25],导致色谱峰产生严重拖尾现象,影响检测结果的准确性.如《规范》[2]2. 1中采用硫酸二甲酯进行衍生化处理的方式测定环吡酮胺. 有文献报道[25]采用强螯合剂EDTA-2K参与竞争,可以阻止环吡酮胺与金属离子产生螯合,改变色谱峰拖尾现象,实现直接测定. 本研究选择实验室常见的EDTA-2Na试剂作为金属离子螯合剂,试验结果表明,采用5 mL乙腈(含0.5%甲酸)提取样品中的待测组分,再用5 mL EDTA-2Na溶液(2 mg/L)稀释定容进行前处理的方法,适用于环吡酮胺及其余49组分的测定.
2. 4 基质效应考察
化妆品基质复杂,样品中的共提取物(例如:胺类、盐类、硬脂酸以及甘油酸酯等)与待测化合物同时从喷雾针流出,将影响目标化合物的雾化、挥发、裂分、化学反应及带电过程,导致发生离子增强或离子抑制的效应,影响分析结果的准确性. 本研究选取水基、乳液和膏霜三种类型的基质对基质效应(matrix effect,ME)进行考察. 分别制备溶剂标准溶液和基质匹配标准溶液,采用基质匹配标准曲线测定比较法考察基质效应. 根据式(1)[2]计算:

式中:Bs表示基质匹配标准曲线的斜率,As表示溶剂标准曲线的斜率. 计算结果正值为基质增强效应,负值为抑制效应. 试验表明,化妆品基质对大多数待分析化合物的测定结果有不同程度的抑制或增强作用(如表3所列). 因此,本文采用基质匹配标准工作曲线法进行定量分析,以降低或消除基质效应对定量分析结果的影响.
2. 5 方法学考察
2. 5. 1 线性范围和检出限
分别配制溶剂和基质匹配标准曲线工作溶液,以50种目标化合物的质量浓度、定量离子峰面积拟合线性回归方程,结果表明所有组分在10~100 ng/mL范围内呈现良好线性关系,相关系数均大于0.991 0.取液态水基化妆品基质空白溶液,添加适量低浓度水平混合标准溶液进行前处理,在信噪比(S/N)分别为3和10时计算方法的检出限和定量限,结果如表3所列. 由表3可见,称样量为0.2 g时,方法检出限为0.05~33 ng/g,定量限为0.15~99 ng/g. 其中40种组分与现行标准相比,检出限与定量限更高,具有更高的灵敏度.
2. 5. 2 加标回收率及重复性
分别在液态水基、乳液及膏霜基质空白样品(不含50种组分)中,按50、100、200 ng/g 3个浓度水平加入50种禁用药物的混合标准溶液,每个浓度水平制备3个平行样,进行加标回收率考察,结果如表4所列. 由表4可见,各组分的平均回收率范围为87.2%~109.7%,相对标准偏差(RSD)不超过10%. 取液态水基空基质白样品6份,添加适量混合标准溶液配制成浓度为50 ng/g的加标样品进行测定,进行重复性考察,RSD值均小于5%,表明重复性良好,结果详见表4.
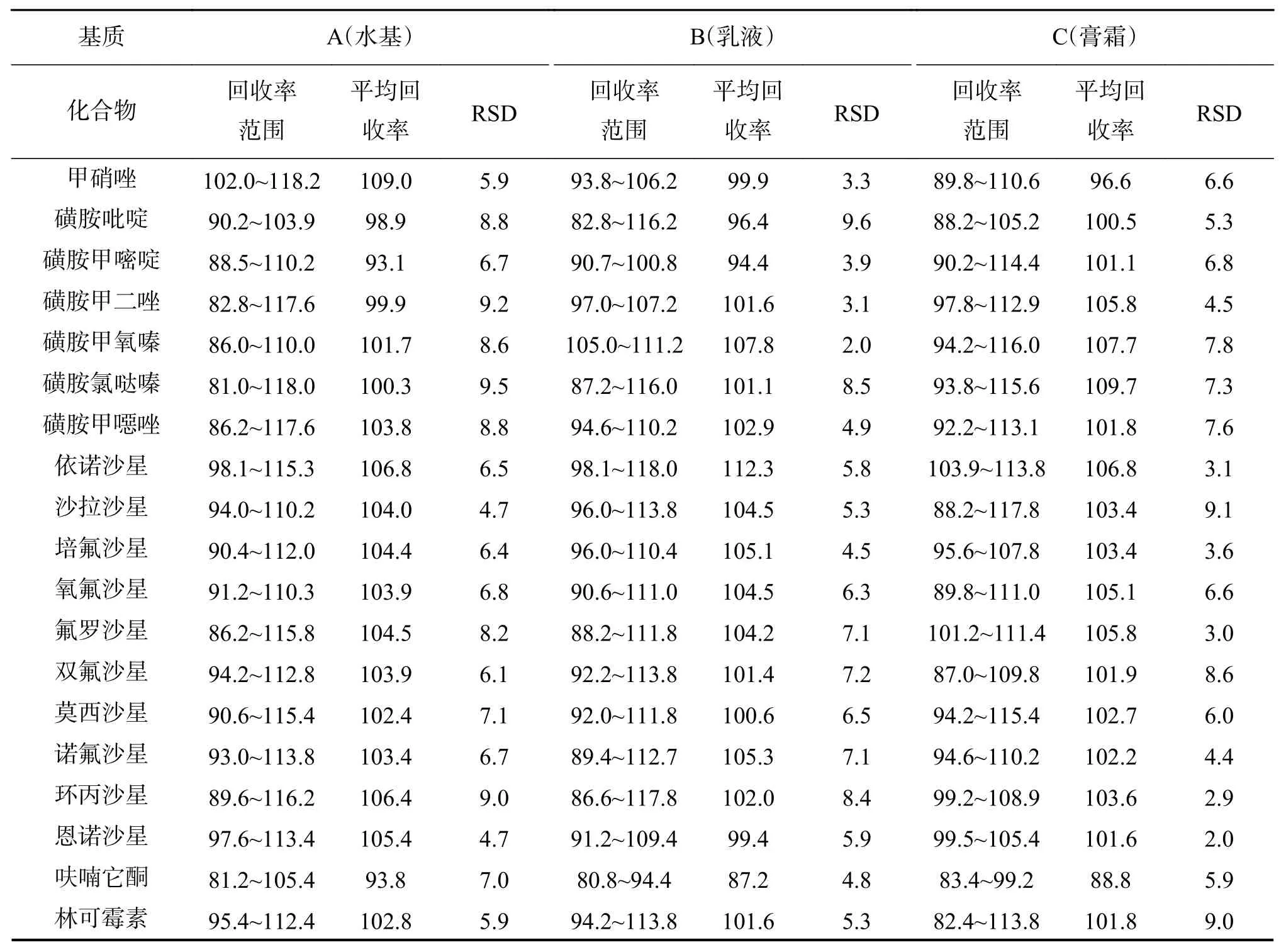
表4 50种化合物的回收率结果Table 4 Recoveries of 50 compounds(%, n=3)

续表 4(%, n=3)
2. 6 样品的测定
应用本文建立的方法对市售30批不同基质的化妆品包括洗面奶(祛痘化妆品)、化妆水、乳液(含儿童、祛痘化妆品)、膏霜(含儿童、祛痘化妆品)及面膜等进行测定,均未检出方法中的50种禁用药物.
3 结论
本文建立UPLC-MS/MS法同时对化妆品中50种非法添加化学药物进行测定. 各组分的线性范围、选择性、重复性和准确性等指标均满足方法学的要求. 对现行标准[4]及《规范》[2]2.1测定方法进行整合完善,优化了样品的制备方法以及流动相分离体系,将现行两个标准所含的40个组分用同一方法进行检测,并拓展性增加了10种常用化学药物的检测,扩大了化妆品中现有标准中禁用组分的筛查范围. 本方法操作简便、重复性好、灵敏度高,
可应用于化妆品中禁用药物的定性筛查和定量分析,为化妆品质量市场监管提供有效的技术参考.

续表 3
猜你喜欢
纺织标准与质量(2022年2期)2022-07-12
现代仪器与医疗(2022年1期)2022-04-19
煤气与热力(2021年12期)2022-01-19
食品安全导刊(2021年20期)2021-08-30
现代仪器与医疗(2021年2期)2021-07-21
科技创新导报(2020年5期)2020-06-11
当代化工(2019年3期)2019-12-12
分析化学(2018年12期)2018-01-22
科教导刊(2017年26期)2017-11-07
科技与创新(2015年17期)2015-09-11
