
发菜盐胁迫响应基因蔗糖合成酶S1的克隆及原核表达
2022-12-29 12:53李永宁陈雪峰刘欢孟广燕王科堂
食品与发酵工业 2022年24期
李永宁,陈雪峰,2*,刘欢,孟广燕,王科堂
1(陕西科技大学 食品科学与工程学院,陕西 西安,710021)2(陕西科技大学 陕西省农产品加工研究院,陕西 西安,710021)
发菜(Nostocflagelliforme)是一种富含营养成分又具有荒漠化防治等生态作用的念珠状蓝藻[1]。野生发菜在干旱、半干旱地区分布居多[2],长期面临低温、干旱、土壤盐渍化等非生物胁迫[3],生命力极强。发菜面临恶劣环境仍能维持自身生长,一方面在于与环境胁迫相关的基因选择性表达产生抵御能力[4-5],另一方面发菜细胞分泌的多糖物质发挥着重要作用[6]。发菜多糖是发菜藻体在生长过程中分泌的黏性长链多糖[7],可使发菜在各种极端环境下具有更好的适应性。同时,大量研究表明发菜多糖亦具有良好的抗炎抑菌效果[8]以及免疫活性[9-10],是一种极具开发潜力的功能性资源[11]。盐胁迫下发菜会积累胞外多糖用于缓解外界不利条件[12],因此,研究发菜盐胁迫响应基因,既可以提高发菜多糖的产量,充分发挥发菜的资源价值,又可以提高发菜耐盐性,提升其环境适应能力。
蔗糖合成酶参与蔗糖代谢等多项生命活动,除去分解蔗糖,还具有参与细胞分化、提高植物抗逆性等多种生物学功能[13]。植物通过消耗蔗糖获取生长养分,面临逆境胁迫时,会积累蔗糖以抵御外界不利环境[14]。因此,开展蔗糖合成酶基因的分子信息及其在模式细胞中表达情况的研究,对明确生物逆境响应的分子机制具有重要意义。
王贵春[15]将发菜在0.3 mol/L盐浓度下培养,以正常组发菜作对照,发现一定盐浓度培养下的发菜胞外多糖质量分数增加了50.3%。赵秀霞[16]利用高通量转录组测序技术(RNA-Seq),对正常培养及盐胁迫(0.3 mol/L NaCl)培养的发菜细胞进行了转录组分析,得到了与发菜多糖合成相关基因(糖基转移酶、UDP-葡萄糖焦磷酸化酶、多糖聚合酶等)的转录水平均发生显著变化的结论。蔡国强等[17]将发菜中的GDP-甘露糖4,6-脱水酶基因进行克隆及表达,论证了发菜合成多糖以响应盐胁迫受到相关基因调控的观点。CUI等[18]将前期发现的耐盐基因drnf1在集胞藻PCC 6803和拟南芥两种模式植株中异源表达,印证了drnf1基因可以在更高水平上上调相关耐盐基因的表达来提高耐盐性的观点。本研究通过PCR技术、基因重组等分子生物学手段克隆蔗糖合成酶S1基因,利用相应软件直观表征目的蛋白氨基酸序列所编码的蛋白质的种类和功能,构建重组表达载体,诱导目的基因表达出预期大小的蛋白质并优化表达条件,为发菜响应盐胁迫相关基因、基因簇、作用位点及其响应机制的研究奠定一定的基础,为从基因水平提高发菜多糖产量提供思路。
1 材料与方法
1.1 材料与试剂
发菜细胞由本实验室用液体BG-11培养基培养[19]。DH5α、BL21感受态细胞及各种试剂盒,天根生化科技(北京)有限公司;pETsumo载体,武汉金开瑞生物工程有限公司。
1.2 仪器与设备
MGC-250P Blue pard光照培养箱,上海一恒科学仪器有限公司;Mastercycler nexus PCR仪,德国Eppendorf公司;YCP-31DN琼脂糖水平电泳仪,艾科仪器设备公司;Genosens 1860电子成像分析系统,山东博科仪器公司。
1.3 实验方法
1.3.1 目的基因TA克隆及序列测定
利用试剂盒提取发菜基因组DNA,质量检测合格后-20 ℃保存待用。
在分析盐胁迫下发菜转录组测序数据的基础上,结合NCBI信息库中所收录的发菜基因组GeneBank信息,比对功能注释和代谢通路,筛选盐胁迫响应基因蔗糖合成酶SS61.1:WP_100899461.1(S1)。利用同源重组的方法,加入NotI酶切位点及其前后部分同源片段设计引物,上游引物序列为S1F:5′-GGTGGTGGATCCGAATTCCGGACTATGTATGAACTGGTTC-AGGCCGTGTTTAACGGTGA-3′,下游引物序列为S1R:5′-CAGTGGTGGTGGTGGTGGTGTTAGCCACCTT-TATGTTTCTCAAGAATTTTTTCGGCACG-3′。按95 ℃,5 min,95 ℃,30 s,55 ℃,1.5 min,72 ℃,1 min,30个循环,72 ℃,7 min的程序进行PCR反应。电泳条带与Marker比对,验证目的基因大小,后将产物回收,连接线性化载体pETsumo,转化DH5α,筛选阳性克隆子培养,抽提质粒后测序验证。
1.3.2 序列分析
将测序结果上传至NCBI数据库中BLAST比对,测序序列与蔗糖合成酶S1序列一致,即表达载体构建成功。依据核苷酸序列推译氨基酸序列,分析目的蛋白的生化性质。在得到目的蛋白氨基酸种类及含量等基础信息后,分析物种间同源性。同时,在ExPASy-ProtScale中依据疏水性标度判别蛋白质的亲/疏水性,在TM-HMM中分析目的蛋白有无跨膜区及所含跨膜区的个数,在NetPhos 3.1 Server中得到目的蛋白的磷酸化位点及个数,便于开展细胞调节通路等方面研究,结合DNAMAN和SPOMA预测信息,推理目的蛋白的二级结构。
1.3.3 蔗糖合成酶基因S1原核表达及表达条件优化
将构建成功的含蔗糖合成酶S1表达载体的BL21菌液(1%)转接到15 mL含卡那霉素(kanamycin,Kan)抗性的无菌LB培养基中,37 ℃、200 r/min条件下过夜培养,活化菌种。抽提重组质粒,转化具有高效表达能力的大肠杆菌BL21细胞,诱导蔗糖合成酶S1蛋白表达,SDS-PAGE检测表达量。
在目的蛋白小量表达验证成功的基础上,对诱导表达的温度、诱导剂诱导时间、加入诱导剂时菌液OD600值、加入诱导剂异丙基-β-D-硫代半乳糖苷(isopropyl-beta-D-thiogalactopyranoside,IPTG)的终浓度进行优化,确定蔗糖合成酶S1的最佳表达条件。
1.3.3.1 IPTG终浓度优化
活化后的菌种(1%)转接到含Kan抗性的LB中培养,当OD600值达到0.6时,加入IPTG使其终浓度分别为0.05、0.1、0.4、1.0 mmol/L,设置空白对照组,继续培养4 h,取菌液制样进行SDS-PAGE,确定诱导剂IPTG最佳终浓度。
1.3.3.2 添加诱导剂时的OD600值优化
按1.3.3.1培养菌种,当OD600值分别达到0.4、0.6、0.8、1.0时,加入IPTG(0.05 mmol/L),继续培养4 h,取菌液制样,SDS-PAGE电泳确定加入诱导剂时所对应的最佳OD600值。
1.3.3.3 添加诱导剂后诱导时间优化
按1.3.3.1培养菌种,当OD600值达到0.8时,加入IPTG(0.05 mmol/L),分别继续培养2、4、6、8、12 h,取菌液制样,SDS-PAGE电泳确定加入诱导剂后最佳诱导时间。
1.3.3.4 诱导温度优化
按1.3.3.1培养菌种,当OD600值达到0.8时,加入IPTG(0.05 mmol/L),分别在16、20、25、30、37 ℃温度梯度下继续培养4 h[20],取菌液制样,SDS-PAGE电泳确定最佳诱导温度。
2 结果与分析
2.1 目的基因 TA克隆
提取发菜基因组DNA,OD260/OD280值为1.83,电泳条带清晰,无杂带(图1),说明DNA质量高,可进行后续实验。蔗糖合成酶S1基因体外扩增在2 400 bp位置出现特异性条带,符合预期(图2)。构建蔗糖合成酶S1基因表达载体pETsumo-S1,转化DH5α,挑阳性克隆扩大培养。菌液PCR结果可见2 900 bp左右条带清晰(图3),结果正确(目的基因片段2 409 bp+克隆位点500 bp),初步验证表达载体构建成功。

M-Marker 15 000+2 000;1~2-发菜DNA图1 发菜基因组DNAFig.1 DNA from N.flagelliforme

M-Marker 15 000+2 000;1-蔗糖合成酶S1基因图2 蔗糖合成酶S1基因PCR扩增Fig.2 PCR amplification of sucrose synthase S1 gene

M-Marker 5 000;1-蔗糖合成酶S1基因菌液PCR鉴定;2-pETsumo空载体PCR图3 蔗糖合成酶S1阳性重组菌菌液PCRFig.3 PCR of sucrose synthase S1 positive recombinant bacteria
2.2 目的蛋白序列分析
2.2.1 核苷酸及氨基酸序列
蔗糖合成酶S1基因纯化回收与线性化载体pETsumo成功连接后,使用载体通用引物对重组质粒双向测序,结果表明克隆得到的目的片段大小为2 409 bp(图4)。分析可知蔗糖合成酶S1蛋白分子式为C4223H6460N1114O1205S23,分子质量大小为92.85 kDa,编码802个氨基酸。该蛋白中含量最高的氨基酸为亮氨酸(Leu)(11.8%),谷氨酸(Glu)(8.5%)次之。蔗糖合成酶S1蛋白的理论等电点为5.52,不稳定系数>40,达到40.84,故蔗糖合成酶S1蛋白为不稳定蛋白。
将蔗糖合成酶S1基因核苷酸序列Blast进行同源性比对(图5),可见该基因保守性较高,与念珠藻(NostoccommuneHK-02)的同源度为93.16%,与念珠藻(Nostocsp. ATCC 53789)的同源度为90.24%,与念珠藻(Nostocsp. C057)的同源度为90.23%,与念珠藻(Nostocsp. TCL240-02)的同源度为89.99%,与念珠藻(NostocedaphicumCCNP1411)的相似度为89.05%。
由氨基酸序列比对分析结果(图6)可知,蔗糖合成酶S1蛋白保守性高,与念珠藻(Nostocsp. C3-bin3)的同源度为95.14%,与念珠藻(Nostocsp. FACHB-892)的同源度为94.01%,与念珠藻(Nostocsp. HG1)的同源度为93.52%,与念珠藻(Nostocsp. FACHB-888)的同源度为93.15%,与念珠藻(Nostocsp. NZL)的同源度为91.02%。
2.2.2 蔗糖合成酶S1基因分析及性能预测
ExPASy-ProtScale检测目的蛋白的疏水性,由图7可知,表明蔗糖合成酶S1蛋白的多肽链中,第271位的缬氨酸正值最大,即疏水性最强,第534位的天冬酰胺负值最大,即亲水性最强。以零值为界上下对比,该蛋白所含的亲水性区间相对疏水区间更多,可知S1蛋白为亲水性蛋白。

图5 蔗糖合成酶S1基因核酸序列系统发育树Fig.5 Phylogenetic tree of sucrose synthase S1 gene sequence

图6 蔗糖合成酶S1基因氨基酸序列系统发育树Fig.6 Phylogenetic tree of sucrose synthase S1 gene amino acid sequence
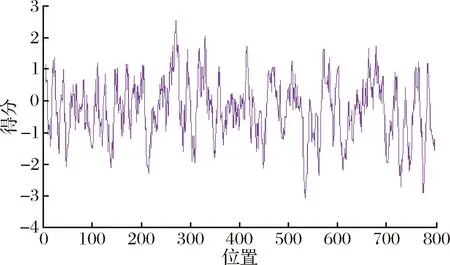
图7 蔗糖合成酶S1(SS61.1)疏水性分析Fig.7 Hydrophilic and hydrophobic analysis of sucrose synthase S1 (SS61.1)
TM-HMM 程序分析目的蛋白跨膜区,结果表明蔗糖合成酶S1蛋白无跨膜区。
NetPhos 3.1分析蔗糖合成酶S1蛋白磷酸化位点,其中包含37个Ser磷酸化位点、13个Thr磷酸化位点以及8个Tyr磷酸化位点。
DNAMAN和SOPMA分析预测蔗糖合成酶S1蛋白的二级结构,结果表明该蛋白含3种二级结构,包括235个α螺旋氨基酸(28.30%),107个β折叠氨基酸(13.34%),460个随机卷曲氨基酸(57.35%)。综合分析结果预测蔗糖合成酶S1蛋白的二级结构主要是α螺旋和随机卷曲。
2.3 蔗糖合成酶S1基因在大肠杆菌中的表达
将构建成功的pETsumo-S1重组表达载体转化大肠杆菌BL21感受态细胞,挑选阳性菌接种到含Kan的LB培养基中过夜培养,后转接(1%)到新鲜LB中至OD600值达到0.6,加入IPTG(终浓度0.1 mmol/L)继续培养。取适量菌液离心弃去上清液,加入电泳缓冲液重悬菌体,于沸水中煮制,SDS-PAGE电泳检测样品。电泳条带显示约109 kDa处有预期大小的外源蛋白表达(图8),其中蔗糖合成酶S1蛋白分子质量为92.85 kDa,His SUMOtag标签为16 kDa,说明S1基因成功诱导表达出目的蛋白。

M-Marker 14.4~116 kDa;1-大肠杆菌蛋白对照;2-0.1 mmol/L诱导剂诱导S1-pETsumo重组蛋白表达图8 蔗糖合成酶S1蛋白诱导表达电泳图Fig.8 Electrophoresis of sucrose synthase S1 protein induction and expression
2.4 蔗糖合成酶S1在大肠杆菌中的表达条件优化
分别对诱导蛋白表达时添加诱导剂的终浓度、添加诱导剂时的OD600值、添加诱导剂后诱导时间和诱导温度进行单因素优化,最终得到蔗糖合成酶S1蛋白的最佳诱导条件为当OD600值为0.8时,添加终浓度为0.05 mmol/L的诱导剂,37 ℃、200 r/min条件下诱导6 h。
2.4.1 诱导剂IPTG终浓度优化
电泳图(图9)泳道1~5分别对应诱导剂IPTG浓度0、0.05、0.1、0.4、1.0 mmol/L,由图9可知在不同终浓度IPTG作用下,蔗糖合成酶S1重组蛋白均有表达,其中0.05 mmol/L终浓度下表达量最高,说明在控制单因素条件下,IPTG终浓度0.05 mmol/L为最适诱导浓度。

M-Marker 14.4~116 kDa;1-不添加诱导剂;2~5-IPTG终浓度分别为0.05、0.1、0.4、1 mmol/L图9 蔗糖合成酶S1重组蛋白诱导表达时IPTG终浓度优化Fig.9 Optimization of IPTG final concentration of sucrose synthase S1 recombinant protein
2.4.2 添加诱导剂时的OD600值
电泳图(图10)泳道1~5分别对应加诱导剂时OD600值为0.8、0.6、0.4、0.2及不加IPTG,由图10可知菌液生长状态不同,即对应不同梯度的OD600值时,加入IPTG(0.05 mmol/L),蔗糖合成酶S1重组蛋白表达量各不相同,OD600值对应0.8时,电泳条带颜色最深,目的蛋白表达量最高,说明在控制单因素条件下,目的蛋白在OD600值为0.8时加入诱导剂更能有效表达。

M-Marker 14.4~116 kDa;1~4-添加诱导剂时OD600值分别为0.8、0.6、0.4、0.2;5-不添加诱导剂图10 蔗糖合成酶S1重组蛋白OD600值优化Fig.10 Optimization of sucrose synthase S1 recombinant protein OD600
2.4.3 添加诱导剂后诱导时间
电泳图(图11)泳道1~5分别对应添加诱导剂后诱导12、8、6、4、2 h,由图11可知蔗糖合成酶S1重组蛋白在不同的诱导时长下表达量有所差异,诱导6 h时目的蛋白表达量最大,说明在控制单因素条件下,IPTG诱导剂诱导6 h最佳。

M-Marker 14.4~116 kDa;1~5-添加诱导剂后分别诱导12、8、6、4、2 h图11 蔗糖合成酶S1重组蛋白诱导剂诱导时间优化Fig.11 Optimization of induction time of sucrose synthase S1 recombinant protein inducer
2.4.4 诱导温度
电泳图(图12)泳道1~5分别对应添加诱导剂后诱导温度为16、20、25、30、37 ℃,由图12可知蔗糖合成酶S1重组蛋白均有表达,诱导温度为37 ℃时,目的蛋白表达量最高,说明在控制单因素条件下,诱导剂IPTG在37 ℃下诱导效果最佳。

M-Marker 14.4~116 kDa;1~5-添加诱导剂后诱导温度分别为16、20、25、30、37 ℃图12 蔗糖合成酶S1重组蛋白诱导温度优化Fig.12 Optimization of induction temperature for sucrose synthase S1 recombinant protein
3 结论与讨论
现阶段,发菜盐胁迫响应基因的研究相对分散,尚未形成完整的分子体系,而高盐胁迫可促进发菜多糖的合成,基因分子信息的丰富对于日后从基因水平提高发菜多糖产量具有重要意义。因此,本研究在筛选盐胁迫响应基因蔗糖合成酶S1的基础上,采用分子生物学方法克隆出长度为2 409 bp的目的基因片段,纯化回收目的片段与pETsumo质粒连接构建表达载体pETsumo-S1,并原核表达出分子质量为92.85 kDa的目的蛋白,优化该蛋白最佳表达条件为菌液在OD600值为0.8时添加终浓度为0.05 mmol/L的诱导剂,37 ℃、200 r/min条件下诱导6 h。这为明确发菜响应盐胁迫与发菜多糖生物合成间的内在联系,系统揭示发菜响应盐胁迫的内在分子调控机制提供理论基础。
然而,阐明发菜响应盐胁迫的调控机理涉及众多基因、基因簇的研究,理论体系庞大,需要筛选并明确生物信息及功能活性的基因数目众多,并且目前表达目的蛋白采用的大肠杆菌宿主细胞与发菜的亲缘关系较远,属于异源表达,对于蛋白功能的验证不足以确保其在蓝藻中有同样的表现。本研究对蔗糖合成酶S1基因氨基酸序列的Blast同源比对分析结果显示,其与多个念珠藻菌株的同源度达到90%以上,可以以此为思路确定模式表达菌株展开今后研究。在模式蓝藻细胞中构建表达通路,直至将重组表达载体导入发菜藻体细胞表达并进行功能验证能够为相关调节机制的研究提供更权威的依据。
猜你喜欢
江苏农业科学(2022年2期)2022-02-15
中国土壤与肥料(2021年5期)2021-12-02
中国甜菜糖业(2020年3期)2020-12-08
北华大学学报(自然科学版)(2020年5期)2020-10-21
疯狂英语·新悦读(2020年7期)2020-07-30
癌变·畸变·突变(2020年1期)2020-02-12
江西农业大学学报(2018年5期)2018-11-22
浙江工业大学学报(2017年5期)2018-01-22
上海医药(2016年23期)2016-12-22
中国糖料(2016年1期)2016-12-01
