
芳基、烯基亚砜与苯酚、苯胺的[3,3]-重排反应进展
2022-12-27 01:06李东阳潘文静
浙江师范大学学报(自然科学版) 2022年1期
彭 勃, 李东阳, 潘文静, 张 磊
(浙江师范大学 化学与生命科学学院,浙江 金华 321004)
[3,3]-重排在有机化学的重排领域具有重要地位,它在天然产物合成与药物合成中有很高的应用价值.21世纪以来,越来越多的有机化学家对[3,3]-重排进行了更深一步的探索[1-17].同时,具有联芳烃化、多官能团化特点的芳基、烯基亚砜[18-22]的重排受到了重点关注.它们与亲核试剂“组装-形成重排前体[23-27]-进行[3,3]重排”的独特反应模式,使反应底物的范围变得更加广泛,更加具有官能团兼容性.苯酚、苯胺作为常见易得的亲核试剂也被应用到亚砜的[3,3]-重排中.通过对该重排机理的研究,发展了控制中间体[28-32],获得双官能团化产物的化学.本文将按照芳基、烯基亚砜与苯酚、苯胺的[3,3]-重排,分类介绍它们的研究进展.
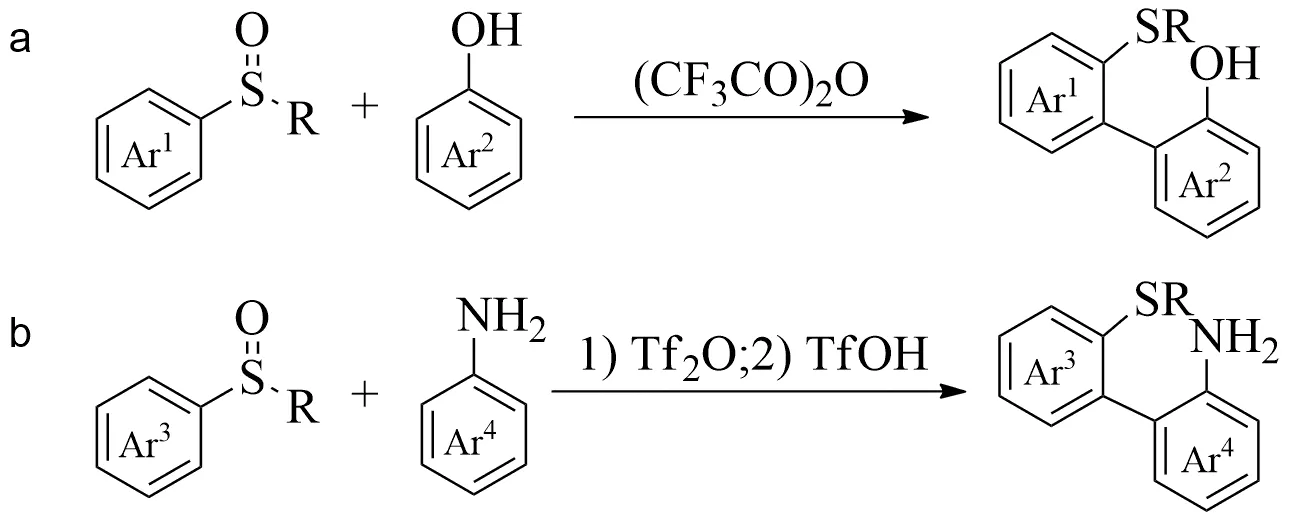
图1 芳基亚砜与苯酚、苯胺[3,3]-重排反应
1 芳基亚砜与苯酚、苯胺[3,3]-重排反应
1.1 芳基亚砜与苯酚、苯胺[3,3]-重排反应简介
2016年,Yorimitsu课题组[33]报道了在三氟乙酸酐(TFAA)作用下,芳基(杂芳基)亚砜与酚类化合物的CAr-CAr偶联反应.反应中,芳基(杂芳基)亚砜经三氟乙酸酐活化后与酚发生亲电组装形成S-O双杂型重排中间体,最后发生Claisen重排形成芳基偶联产物(见图1a).该反应为无金属介入的基于亲电重排的C-C偶联反应,且反应条件温和,对芳基亚砜和杂芳基亚砜底物都有很好的适用性,同时对酚类底物也具有很广的适用范围.2020年,Yorimitsu等课题组[34]又报道了芳基亚砜与苯胺的重排反应.在用三氟甲磺酸酐活化亚砜的基础上,加入三氟甲磺酸与组装后的中间体反应,形成重排前体.然后,脱氢恢复芳香性即可得到重排产物(见图1b).
1.2 芳基亚砜的底物范围研究
在最优反应条件下,Yorimitsu等课题组[33]首先对芳基亚砜的适用范围进行一系列探究.发现用三氟乙酸酐在二氯甲烷中处理吲哚亚砜和苯酚,在25 ℃下得到3-(2-羟苯基)-2-甲硫基-1-(4-甲苯磺酰基)吲哚1a,产率为89%(见图2).3-(2-羟苯基)-2-甲硫基-苯并呋喃1b可以大量制备,也说明了该反应的稳定与实用性.在苯基甲基亚砜的3号位引入氟原子时,重排产物则表现出一定的区域选择性,产率比1e∶1f=5∶1.当位阻增大,在3号位引入三异丙基硅醚基时,便得到单一的位阻小的重排产物(1g).当亚砜的硫原子在环内时,如苯并噻吩亚砜[35-37],改变反应条件,同样能够发生[3,3]重排,得到3-(2-羟基苯基)苯并噻吩1c.同时由于原料与产物的化学性质的巨大差异,使得产物容易制备和分离,产率为79%~93%.
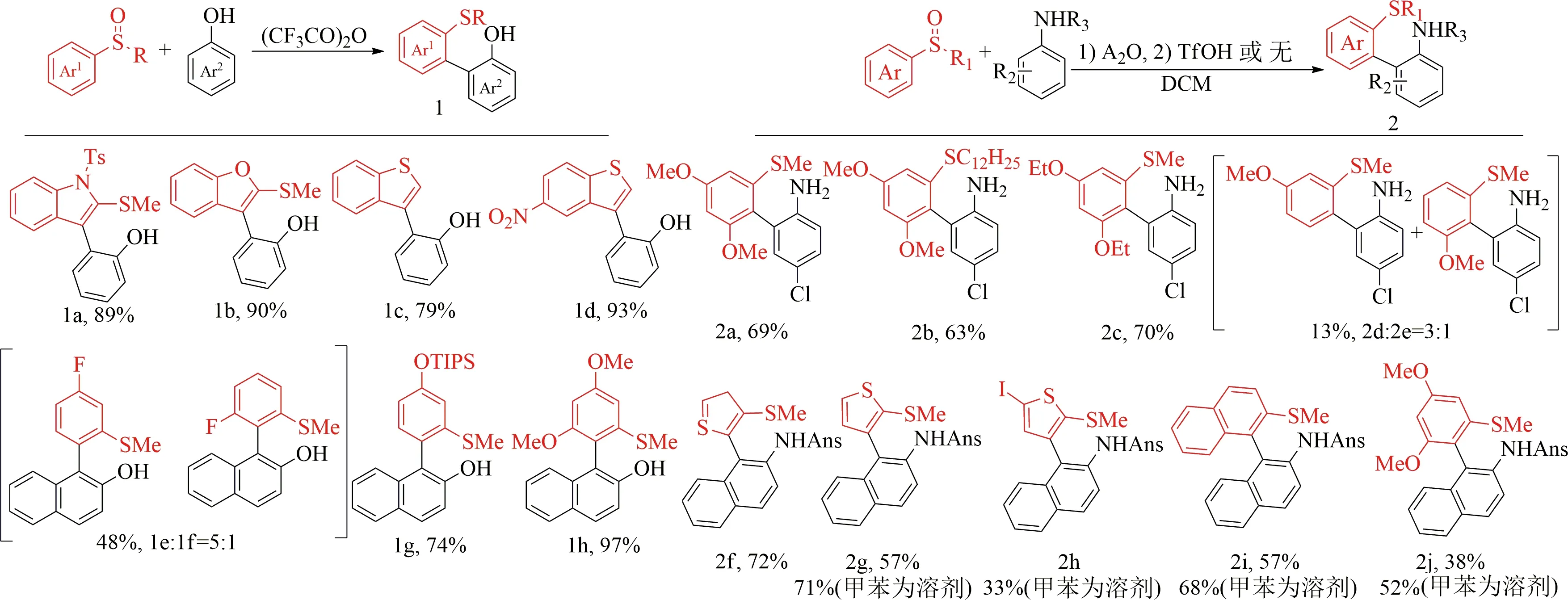
图2 芳基亚砜的底物范围
芳基亚砜与苯胺的[3,3]-重排反应(见图2),分2种情况:1)加入三氟甲磺酸稳定苯胺N上的质子.此时,芳基亚砜间位的供电子基起着重要的作用.含3,5-二烷氧基苯基甲基的亚砜具有良好的产率69%(2a).当苯基的间位只有1个供电子基时,产率急剧下降至13%(2d,2e);而当苯基的间位没有供电子基时,便无法得到重排产物[34].2)引入保护基.当以烷基磺酰基(Ans)为保护基团时,杂芳族硫化物进行了有效的反应,得到了良好的产率.芳香族2-萘基亚砜和3,5-二甲氧基苯基亚砜分别与Ans保护的2-萘胺反应,生成2i和2j,产率分别为57%,38%.在2g—2j的合成中,甲苯是比二氯甲烷更好的溶剂,但溶剂效应的原因尚不清楚[38].
1.3 苯酚的底物范围研究
在苯酚[33]的对位无论引入供电子基(叔丁基、甲氧基),还是吸电子基(醛基、三氟甲基、溴)都能得到相应产物3a—3e的较高产率77%~97%(见图3).在2-碘苯酚重排后,得到单一的邻位产物3f.而3-溴苯酚在重排过程中则表现出一定的区域选择性,在溴的对位和邻位均能得到重排产物3h和3i,具有91%的产率,产率比为2.6∶1.0.

图3 苯酚的底物范围
1.4 苯胺的底物范围研究
苯胺对位的吸电子基团在苯胺与苯基烷基亚砜中具有关键性作用[34].卤素、酯、三氟甲基和硝基(4a—4d),都能取得60%~80%良好的产率(见图4).而供电子较强的官能团如甲氧基,则没有反应.在用2-甲亚磺酰基苯并噻吩代替3,5-二甲氧基苯基甲基亚砜后,对苯胺的几个保护基进行了考察[38],发现磺酰基保护具有积极的作用.其中,对甲氧基苯磺酰基保护的苯胺产率最高,达到86%(4i),而对甲基、氰基苯磺酰基与甲基磺酰基保护的苯胺重排效果降低(4f—4h).

图4 苯胺的底物范围
1.5 芳基亚砜与苯酚、苯胺[3,3]-重排反应机理
苯酚与酸酐活化的芳基亚砜亲核组装[33]首先形成重排前体.而后芳基亚砜与苯酚同时去芳香化发生[3,3]-重排,新的C-C键形成.最后双双恢复芳香性,得到二芳基偶联产物(见图5).该二芳基产物具有酚羟基和烷硫基双官能团,具有很高的衍生化应用价值.此时如果不加入三氟甲磺酸,则体系里的三氟甲磺酸根发挥碱的作用,拔掉N上的H,得到一种硫基亚胺的物质;如果加入三氟甲磺酸,则N被质子化发生去芳香化[3,3]或[5,5]重排,同时由于甲氧基的供电子效应,重排产物得以稳定.最后苯环恢复芳香性得到最终产物.

图5 芳基亚砜与苯酚、苯胺 [3,3]-重排反应机理

图6 烯基亚砜与苯酚、苯胺[3,3]-重排反应
2 烯基亚砜与苯酚、苯胺[3,3]-重排反应
2.1 烯基亚砜与苯酚、苯胺[3,3]-重排反应简介
2010年,Yorimitsu等课题组[39]设计了一条简洁的、以多样性为导向的2-甲硫-3-三氟甲基-苯并呋喃的合成路线.各种酚类与被酸酐活化的烯基亚砜发生扩展Pummerer环化反应,直接转化为相应的2-甲基硫-3-三氟-甲基苯并呋喃(见图6a).产物的甲基硫基团经历了进一步的转变,可增加三氟甲基苯并呋喃的多样性.2020年,Yorimitsu等课题组[40]又开发了一种基于N-N的Fischer吲哚合成的S-N变异体.用强力酸酐处理芳基和甲硫基取代的烯基烷基亚砜,然后与有保护基的苯胺反应,得到一种吲哚(见图6b).初始中断的Pumerrer反应将在原位产生关键的S-N系前体,然后进行[3,3]-重排,最后在遵守Fischer规则下形成吲哚环.
2.2 烯基亚砜的底物范围研究
在TFAA存在下[39],用苯基甲硫基取代的乙烯基甲基亚砜与4-叔丁基苯酚反应,得到相应的苯并呋喃5c(见图7),产率87%.未取代的和甲基取代的甲硫乙烯基甲基亚砜5a—5b,获得67%和78%的产率.在烯基亚砜上引入各种取代基的反应也是成功的(5e—5g),如吸电子基团(4-三氟甲基苯基)、给电子基团(4-甲氧基苯基)和卤素(4-氯苯基).体积较大的1-萘基和杂芳基(噻吩)取代的烯基亚砜,也具有同样较高的产率85%和78%(5d和5h).
以甲苯为溶剂[40],在-40 ℃下用五氟丙酸酐处理对甲基氧基苯磺酰基保护的对甲基苯胺和对叔丁基苯基取代的烯基亚砜,反应15 h,得到了87%的吲哚衍生物6b(见图7).包括对位取代的氰基和酯基在内的一系列芳基也被发现是相容的(6d—6f).强电子供体对甲氧基将显著影响酸酐活化烯基亚砜物种的稳定性和反应性,从而形成复杂的混合物6g.而间甲氧基不应影响相应亚砜物种的性质,可以得到80%的产率(6h).
2.3 苯酚的底物范围研究
对位取代的酚类与三氟甲基取代的烯基亚砜反应时[39],对位有吸电子基团的酚类(如氰基、三氟甲基、酯基)亲核性较弱,反应需要加热到40 ℃的温度才能达到较高的产率(7a—7c)(见图8).
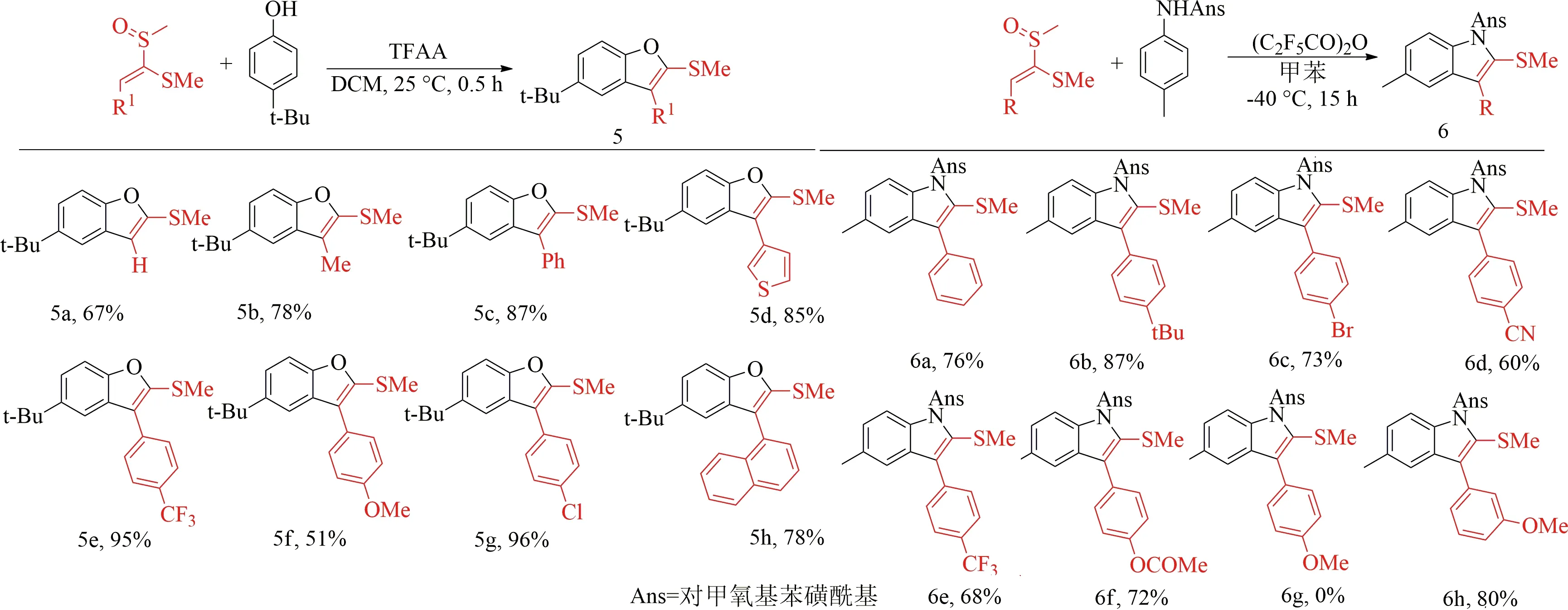
图7 烯基亚砜的底物范围
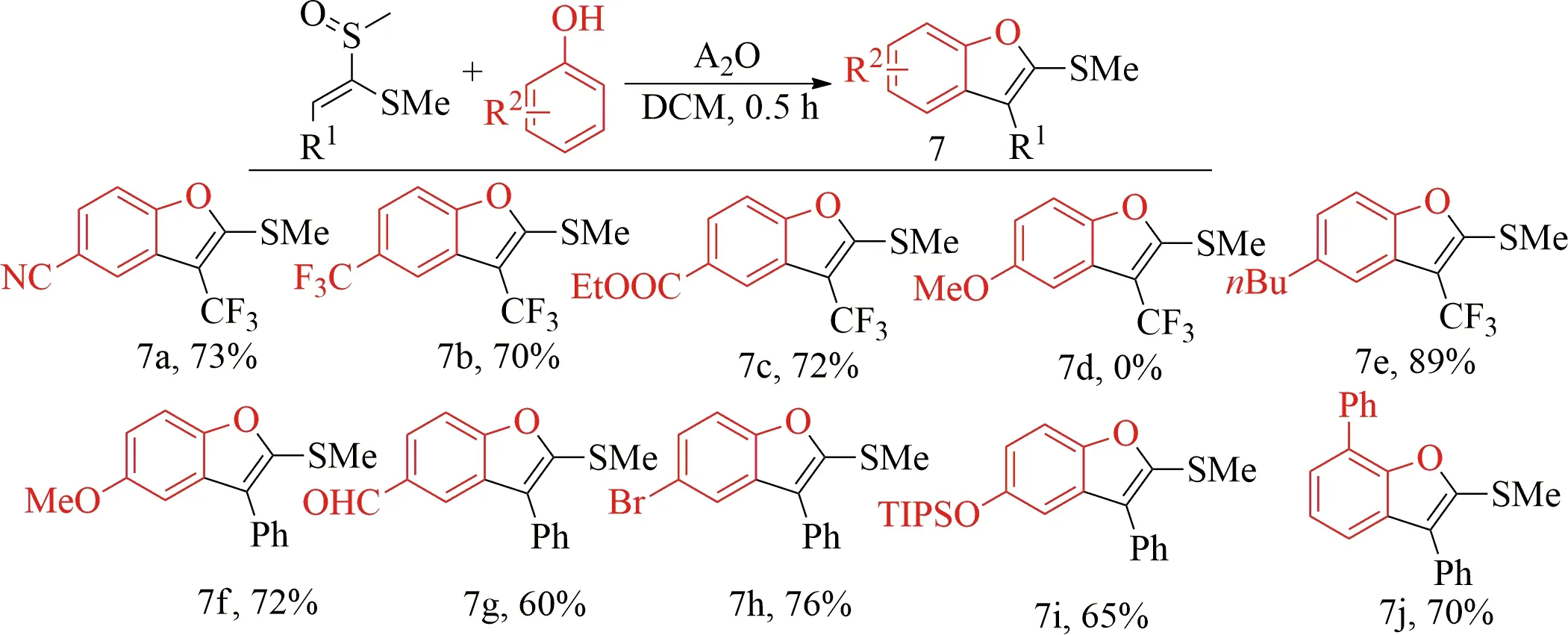
图8 苯酚的底物范围
相反,对甲氧基苯酚被证明太活泼,在-40 ℃下依然不能转化为相应的苯并呋喃7d.而对正丁基苯酚,由于给电子能力相对较弱,在0 ℃下即可得到89%产率(7e).对位取代的酚类与苯基取代的烯基亚砜反应[41],则显示出较好的官能团兼容性,能够与具有卤素、吸电子基或供电子基的酚类物质顺利反应(7f—7h).但醛基在此时的反应条件不能得到重排产物,因为反应过程中产生的三氟乙酸催化羰基与甲硫醇进行二硫缩醛化.加入K2CO3可以抑制这一副反应,从而得到呋喃产物7g,产率为60%.位阻较大的对位三异丙基硅醚基取代的苯酚和邻位苯基取代的苯酚可以分别得到65%和70%的产率(7i和7j).
2.4 苯胺的底物范围研究
同苯酚与烯基亚砜时所表现的性质类似[40],对位有给电子基团的苯胺(如甲氧基)亲核性较强,反应需要冷却到-78 ℃,才能稳定得到58%的产率(8a)(见图9).而对位没有给电子基团的苯胺则只需冷却到-20 ℃,即可得到58%的产率(8b).同时,延长反应时间,也有利于产物的形成.将8c的反应时间由15 h延长到40 h后,得到的8d产物的产率也由原来的56%增加到76%;保护基团不同,其反应效果也不同.三氟甲基磺酰基比对甲氧基苯磺酰基的保护效果更好,以硝基甲烷为溶剂,与室温下三氟乙酸酐活化的亚砜反应,产率由原来的0%(8e)增加到58%(8f).
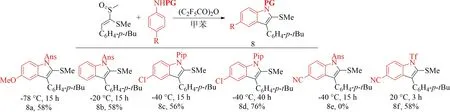
图9 苯胺的底物范围
2.5 烯基亚砜与苯酚、苯胺[3,3]-重排反应的机理研究
在Tf2O活化烯基亚砜后[39],酚羟基亲核进攻硫阳离子,产生重排前体.然后经历快速的[3,3]-重排,在苯酚的邻位处形成新的碳碳键.接下来环化得到二氢苯并呋喃,最后在三氟甲磺酸的存在下发生甲硫醇消除反应(见图10).烯基亚砜与苯胺的反应机理[40]与烯基亚砜与苯酚不同的是N上需要保护集团,否则重排前体不会发生[3,3]-重排,而是在三氟甲磺酸根的作用下脱去H,从而形成N=S双键,具体可参考芳基亚砜与苯胺的反应机理(见图10).

图10 烯基亚砜与苯酚、苯胺[3,3]-重排反应的机理
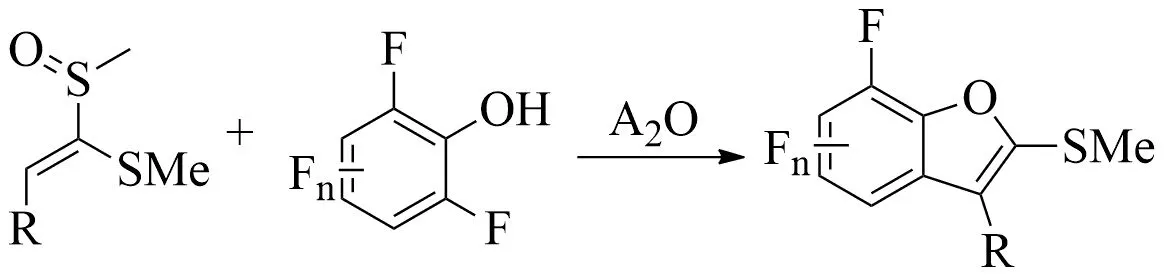
图11 亚砜与多氟取代苯酚[3,3]-重排反应
3 亚砜与多氟取代苯酚[3,3]-重排反应
3.1 亚砜与多氟取代苯酚[3,3]-重排反应简介
由于氟的体积小、电负性高等独特的性质,使含氟化合物在药物、农用化学品和光电子领域引起了越来越多的关注.因此,有机化学家们作出了大量的努力,开发各种方法以有效和选择性地合成含氟化合物.2018年,Yorimitsu课题组[30]突破经典SNAr方法合成氟化物对底物范围的限制,发展出了以[3,3]-重排为核心的合成氟化物新方法(见图11).
3.2 亚砜的底物范围研究
萘环的低芳香性会促进酚羟基进攻活化之后的亚砜,提高[3,3]-重排的效率[31].杂芳基亚砜(如2-噻吩基、2-呋喃基和2-吡咯烷基)与全氟萘酚反应,获得了良好的产率67%~78%(9a—9c)(见图12).而且在过渡金属催化下,噻吩亚砜上的碘基也表现出很好的耐受性.除杂芳基砜外,2-萘亚砜也能克服位阻效应,可以得到85%的产率(9f).而苯基亚砜不适用于此类脱氟重排反应,可能是由于苯环的芳香性较强,反应后仅观察到原料和相应的硫化物.这意味着杂芳基和萘基亚砜的芳香性弱于苯基亚砜的芳香性,这对于重排时的去芳香化是至关重要的.
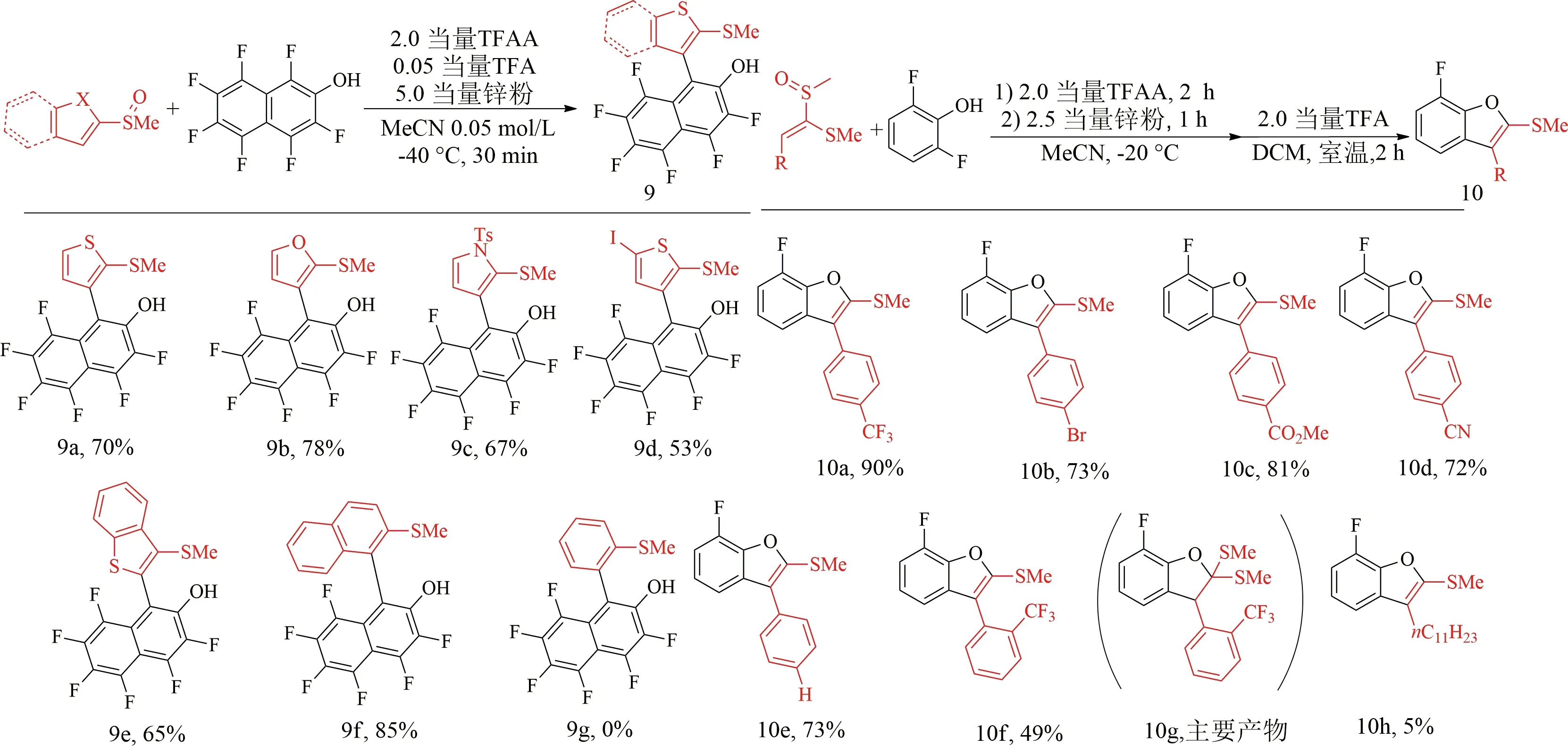
图12 亚砜的底物范围
缺电子的烯基亚砜和电中性的烯基亚砜都能够与2,6-二氟苯酚顺利反应[30],成功得到相应的苯并呋喃10a—10f(见图12).值得注意的是,在反应过程中,烯基亚砜上的溴、氰基和酯基基团都能够很好地兼容10b—10d.当烯基亚砜的苯基邻位有三氟甲基取代时,产物主要是二氢苯并呋喃10g.而使用更有效的脱甲硫基的水合TsOH代替TFA,在甲苯中加热到110 ℃,反应1 h后,可得到苯并呋喃产物10f.烷基取代的烯基亚砜不适于生成呋喃产物10h,这是由于第一步S-O键的组装与后来发生的重排在该情况下很难进行.
3.3 亚砜与多氟取代苯酚[3,3]-重排反应的机理研究
在酸酐存在下[31],由多氟苯酚和芳基亚砜建立S-O键,[3,3]-重排之后得到中间体,它可以通过Zn介导的脱氟恢复芳香性转化为所需的联芳烃(见图13).该反应在酸酐的辅助下以阻断Pumerrer反应的方式组装[30],生成重排前体.随后[3,3]-重排,得到去芳香化的中间体.随后在温和的条件下发生还原脱氟,并以恢复芳香性为驱动力.在环化和消除甲硫醇之后,得到所需的氟化苯并呋喃(见图13).

图13 芳基、烯基亚砜与多氟取代苯酚[3,3]-重排反应的机理
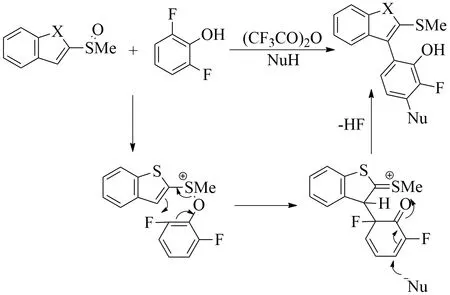
图14 苯酚双官能团化
3.4 苯酚双官能团化
芳基亚砜与多氟取代苯酚重排之后的中间体是2,4-环己二烯酮的结构,具有相当活泼的反应性.2020年,Yorimitsu等课题组[32]将该中间体用于迈克尔加成反应.与邻醌单缩醛的反应相似,亲核试剂进攻中间体的3号位,然后脱掉HF,恢复芳香性(见图14).
3.5 双官能团化的多氟取代苯酚底物范围研究
在2,6-二氟苯酚和2-苯并噻吩亚砜中生成去芳香化中间体后[32],加入哌啶(6.0当量),使溶液加热至室温,搅拌1 h,得到双官能团化氟苯酚11a,产率为71%(见图15).其中过量的碱用于中和反应中产生的三氟乙酸.除了哌啶,二乙胺也可以作为亲核试剂,得到双官能团化产物11b,产率68%.当以伯胺为亲核试剂时,中间体也可以发生加成反应,以37%的产率得到11f.而苯胺由于亲核性较弱,仅有24%的产率(11e).环己胺分别与4-氯-2,6-二氟苯酚和4-碘-2,6-二氟苯酚反应,以中等产率得到相应的氟酚11g—11h.
4 总 结
本文介绍了芳基、烯基亚砜与苯酚、苯胺的[3,3]-重排反应,合成联芳烃、苯并噻吩、苯并呋喃及双官能团化产物的方法.发展出了独特的“组装-重排”模式,该方法具有良好的官能团兼容性且产率优良.其中亲核试剂由苯酚向苯胺的过渡,更是通过质子化或引入保护基的方法,实现了[3,3]-重排.通过捕获重排后的中间体,使用亲核试剂进行迈克尔加成,取得了对多氟取代苯酚双官能团化的效果,体现了该反应模式在[3,3]-重排反应中的应用价值.

图15 多氟取代苯酚的底物范围
猜你喜欢
含能材料(2022年10期)2022-10-22
化学与生物工程(2022年9期)2022-09-30
环境工程技术学报(2022年3期)2022-06-05
化工设计通讯(2022年4期)2022-04-28
能源化工(2021年6期)2021-12-30
能源化工(2021年6期)2021-12-30
纺织检测与标准(2021年3期)2021-12-03
合成技术及应用(2021年1期)2021-01-07
石油化工自动化(2020年1期)2020-03-05
中国循证儿科杂志(2019年2期)2019-06-04
