
炒火麻仁饮片标准汤剂特征图谱及有效成分的定量检测
2022-12-26 03:57:30史紫娟林碧珊陈锦霞刘勇高永坚区淑蕴曾杉
中国现代中药 2022年11期
史紫娟,林碧珊,陈锦霞,刘勇,高永坚,区淑蕴,曾杉
国药集团 广东环球制药有限公司,广东 佛山 528305
火麻仁又名大麻仁、麻子仁、线麻子、大麻子等,为桑科1 年生草本植物大麻Cannabis sativaL.的干燥成熟果实或成熟去壳的种子。炒火麻仁为火麻仁清炒后的炮制品,具有润肠燥、滋阴血之功效[1]。炒火麻仁的主要活性成分有脂肪油、生物碱等。脂肪油为其主要药效成分之一,包含饱和脂肪酸、不饱和脂肪酸及其酯类等,具有降血压、利尿、镇痛、抗炎、抗血栓形成的功效[2]。炒火麻仁中含有的主要生物碱成分为胡芦巴碱,是一种广泛分布的季铵盐生物碱。现代药理学研究表明,胡芦巴碱具有抗肿瘤、降低胆固醇及降血糖等药理活性[3],被视为炒火麻仁的特征性指标成分。关于炒火麻仁的成分含量测定的文献报道不多,现有文献只针对火麻仁中的脂肪酸或胡芦巴碱进行单方面研究[4-5],但由于样品前处理复杂和高温下易发生双键异构化而难以准确测定等原因,《中华人民共和国药典》(以下简称《中国药典》)2020 年版尚未建立炒火麻仁的含量测定指标。
中药饮片标准汤剂是在中医理论指导下以临床实际应用为参考,使用现代中药提取方法并通过标准化工艺规程制备而成的单味中药饮片水煎液,是中药配方颗粒生产工艺和质量的“基准”[6]。本研究制备了20 批炒火麻仁饮片标准汤剂,考察了炒火麻仁饮片标准汤剂的出膏率、总脂肪酸和葫芦巴碱含量及转移率,并进行特征图谱相似度研究,为炒火麻仁配方颗粒的质量控制提供参考。
1 材料
1.1 仪器
H-Class 型超高效液相色谱仪(美国Waters 公司);MS105DU 型十万分之一天平、AL104 型万分之一天平、PL203 型千分之一天平(瑞士梅特勒-托利多公司);KQ-500DE 型数控超声波清洗器(昆山市超声仪器有限公司);FTS-40F 型陶瓷保健壶(潮州市一壶百饮电器实业有限公司);TRL-0.5 型真空冷冻干燥机(大连双瑞科技有限公司);Synergy UV型超纯水仪(美国Millipore公司)。
1.2 试药
对照品α-亚麻酸(批号:111631-201605,纯度:99.8%)、亚油酸(批号:111622-201203,纯度:99.1%)、鸟苷(批号:111977-201501,纯度:93.6%)、胡芦巴碱(批号:110883-201604,纯度:78.4%)均购自中国食品药品检定研究院;色谱纯甲醇和乙腈;色谱纯冰乙酸和十二烷基磺酸钠(Aladdin公司);石油醚为分析纯;水为超纯水。
1.3 样品
20 批火麻仁药材分别产自山西、河南、陕西、甘肃等地,经江阴天江药业有限公司唐波研究员鉴定为桑科植物大麻Cannabis sativaL.的干燥成熟果实,按《中国药典》2020 年版标准检验全部合格,参考《中国药典》2020 年版火麻仁项下炮制方法制成炒火麻仁饮片[1],具体炮制方法:除去杂质和果皮,文火清炒至微黄。依据“逢籽必破”原则,用中药饮片机将炒火麻仁饮片打碎至全部通过10 目筛。20批饮片信息见表1。
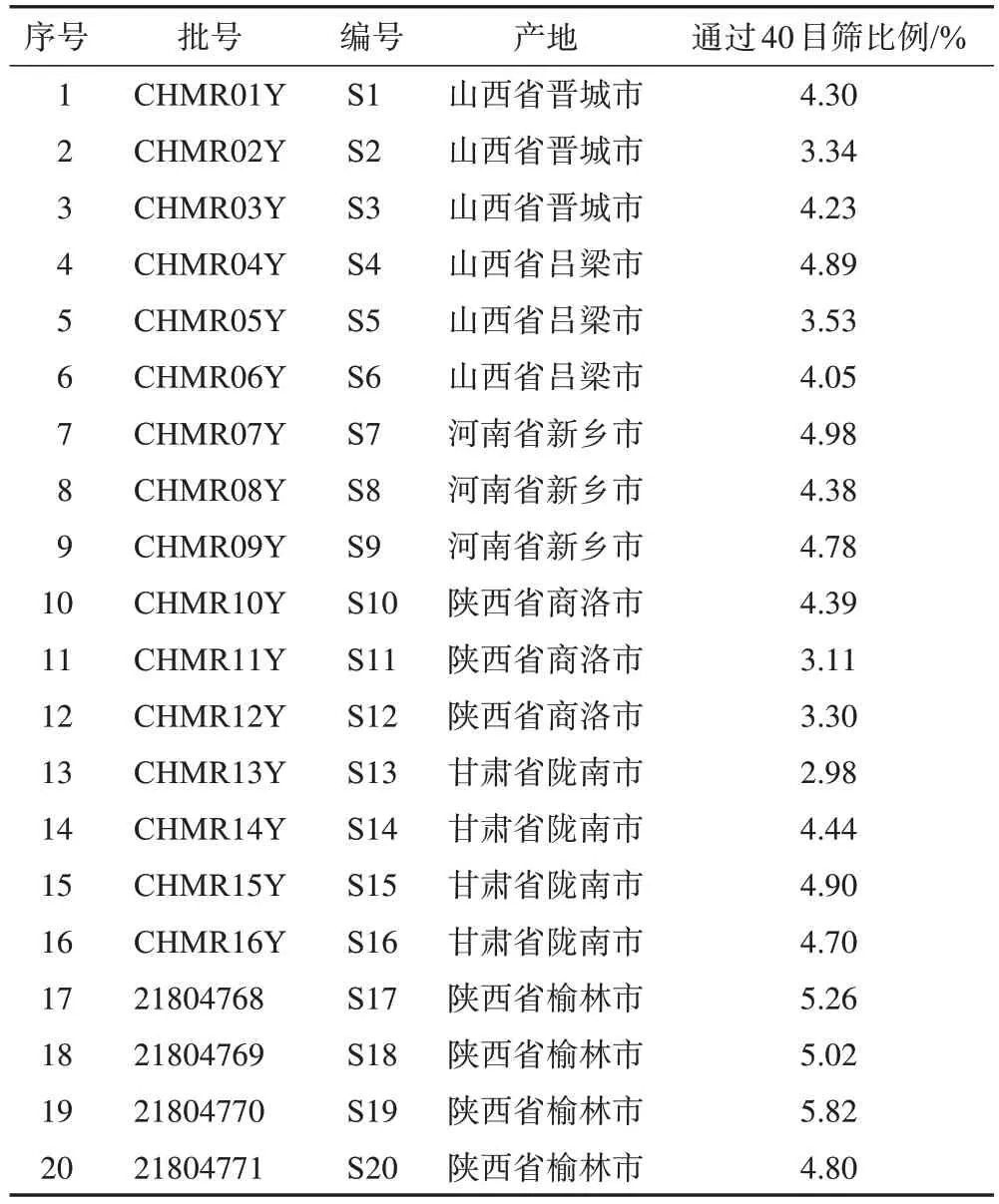
表1 20批炒火麻仁饮片信息
2 方法与结果
2.1 炒火麻仁饮片标准汤剂的制备
取炒火麻仁饮片100 g,加入8倍量水浸泡30 min,煎煮30 min,趁热滤过,药渣再加入6 倍量水,煎煮25 min,滤过,合并滤液。减压浓缩至约200 g的浸膏,即得炒火麻仁饮片标准汤剂。浸膏加水稀释至固含率约为15%,再进行冷冻干燥,即得炒火麻仁饮片标准汤剂冻干粉。
2.2 炒火麻仁饮片和标准汤剂中有效成分的定量测定
《中国药典》2020 年版火麻仁药材、饮片及炒火麻仁饮片均没有相应的含量测定项,本研究参考文献[7-8],采用油重法测定炒火麻仁饮片和标准汤剂中总脂肪的含量。另参照《中国药典》2020 年版(一部)胡芦巴项下胡芦巴碱含量测定色谱条件建立了炒火麻仁饮片和标准汤剂中胡芦巴碱的含量测定方法,为制定炒火麻仁配方颗粒的质量控制标准提供参考[1]。
2.2.1 总脂肪含量测定
2.2.1.1 饮片供试品的制备 称取打碎后炒火麻仁饮片(过二号筛)约2.0 g,移入滤纸筒内,将滤纸筒放入索氏提取器的抽提筒内,由提取器冷凝管上端加入石油醚至接收瓶内容积的1/2处,于水浴上加热,使石油醚不断回流抽提至6 h。将抽提液减压浓缩,分别转移至蒸发皿,置水浴锅上蒸干,再于100 ℃烘箱中干燥1 h,于干燥器内冷却0.5 h 后称质量,蒸发皿增加的质量即为总脂肪质量。
2.2.1.2 标准汤剂供试品的制备 取炒火麻仁饮片标准汤剂冻干粉约1.0 g,以下与2.2.1.1 项下“移入滤纸筒内”后操作相同,蒸发皿增加的质量即为总脂肪质量。
2.2.1.3 标准汤剂总脂肪含量平行样验证 取同一批炒火麻仁饮片标准汤剂冻干粉,平行3 份,按2.2.1.2项下方法制备供试品,测定总脂肪含量。结果3 批标准汤剂的总脂肪质量分数分别为32.08%、32.75%、31.89%,均值为32.24%,RSD为1.40%。结果表明,同一批炒火麻仁饮片标准汤剂平行样总脂肪含量的RSD<2%,表明各平行样间差异较小,标准汤剂制备工艺可靠,总脂肪含量测定方法稳定。
2.2.2 胡芦巴碱含量测定
2.2.2.1 色谱条件与系统适用性试验 ACQUITY UPLC HSS T3 色谱柱(100 mm×2.1 mm,1.8 μm),以甲醇-0.05%十二烷基磺酸钠溶液-冰乙酸(20.0∶80.0∶0.1)为流动相,检测波长为265 nm,柱温为30 ℃,流速为0.30 mL·min-1,进样量为1 μL,理论板数按胡芦巴碱峰计算应不低于4000。
2.2.2.2 对照品溶液的制备 取胡芦巴碱对照品适量,精密称定,加50%甲醇制成质量浓度为20 μg·mL-1的溶液,即得。
2.2.2.3 饮片供试品溶液的制备 取炒火麻仁饮片粉末(过二号筛)约1.0 g,精密称定,置具塞锥形瓶中,精密加入50%甲醇25 mL,密塞,称定质量,超声处理(500 W,40 kHz)30 min,取出,放冷,再称定质量,用50%甲醇补足减失的质量,摇匀,滤过,取续滤液,即得。
2.2.2.4 标准汤剂供试品溶液的制备 取本品冻干粉约0.2 g,精密称定,以下与2.2.2.3 项下“置具塞锥形瓶中”后相同操作,取续滤液,即得。
2.2.2.5 方法学考察 线性关系考察:精密称取胡芦巴碱对照品12.23 mg,置100 mL量瓶中,加50%甲醇溶解并定容至刻度,作为对照品储备溶液。分别精密量取上述储备液,稀释成质量浓度为0.006 1、0.012 2、0.024 5、0.048 9、0.097 8、0.122 3 mg·mL-1的对照溶液,按2.2.2.1项下色谱条件进行测定,记录色谱峰面积。以胡芦巴碱峰面积为纵坐标(Y),质量浓度为横坐标(X),绘制标准曲线,回归方程:Y=168.35X+0.012 3(r=1.000 0),表明质量浓度为0.006 1~0.122 3 mg·mL-1时与峰面积线性关系良好。
精密度试验:取胡芦巴碱对照品溶液,按2.2.2.1 项下色谱条件连续进样6 次,计算胡芦巴碱峰面积的RSD为0.4%,表明仪器精密度良好。
稳定性试验:取炒火麻仁饮片标准汤剂冻干粉约0.2 g,精密称定,按2.2.2.4 项下方法制备供试品溶液,精密吸取供试品溶液,按2.2.2.1 项下色谱条件,分别在0、2、6、12、24 h进样,计算胡芦巴碱峰面积的RSD为1.1%,表明供试品溶液在24 h内稳定性良好。
重复性试验:取同一批炒火麻仁饮片标准汤剂冻干粉约0.2 g,精密称定,按2.2.2.4 项下方法平行制备6 份供试品溶液,精密吸取供试品溶液,按2.2.2.1项下色谱条件测定,计算供试品溶液中胡芦巴碱含量的RSD为0.8%,表明该方法重复性良好。
加样回收率试验:取同一批已知含量的炒火麻仁饮片标准汤剂冻干粉约0.1 g,平行称取6 份,精密称定,置圆底烧瓶中,分别按1∶1 加入胡芦巴碱对照品溶液,按2.2.2.4 项下方法制备供试品溶液,精密吸取供试品溶液,按2.2.2.1 项下色谱条件测定,计算胡芦巴碱平均回收率和RSD,结果胡芦巴碱的平均回收率为97.7%,RSD 为0.3%,表明该方法回收率良好。
2.2.2.6 炒火麻仁饮片标准汤剂出膏率测定 将20批炒火麻仁饮片制备成标准汤剂,取标准汤剂约10 g,置质量恒定的蒸发皿中,水浴蒸干,再放置105 ℃烘箱中干燥3 h,取出,置于干燥器中放冷30 min,测定干固物并按公式(1)计算出膏率。结果见表2。结果表明,20 批炒火麻仁饮片标准汤剂出膏率为15.55%~22.04%,均值为18.80%。

2.2.2.7 炒火麻仁饮片和标准汤剂样品测定 根据已建立的含量测定方法对20批炒火麻仁饮片及其标准汤剂进行定量测定并按公式(2)、(3)计算转移率,结果见表2。
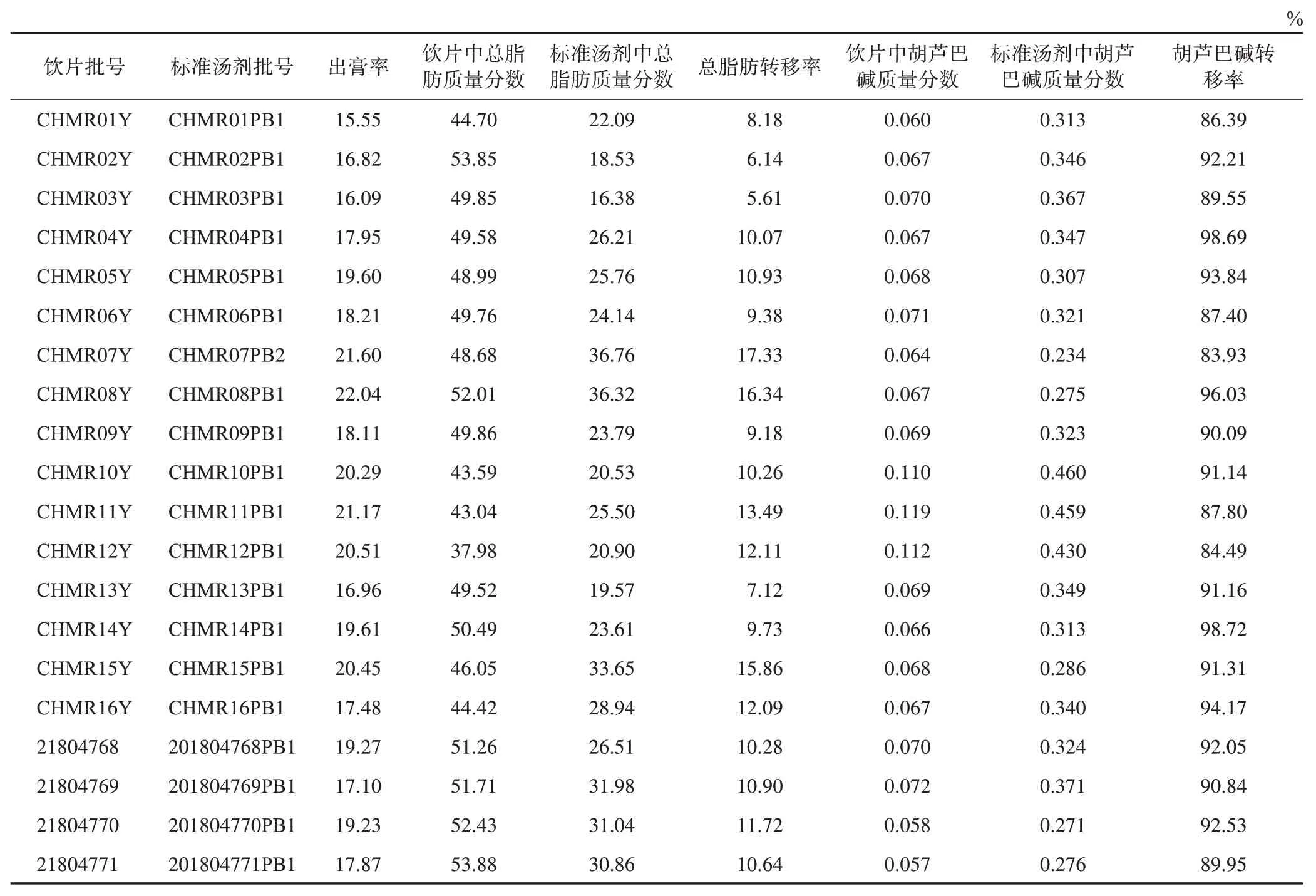
表2 炒火麻仁饮片及标准汤剂测定结果

结果表明,20 批炒火麻仁饮片总脂肪质量分数为37.98%~53.88%,均值为48.58%,;20 批炒火麻仁饮片标准汤剂总脂肪质量分数为16.38%~36.76%,均值为26.15%;20批炒火麻仁饮片标准汤剂总脂肪含量转移率为5.61%~17.33%,均值为10.87%。
20 批炒火麻仁饮片中胡芦巴碱质量分数为0.057%~0.119%,均值为0.074%;20批炒火麻仁饮片标准汤剂胡芦巴碱质量分数为0.234%~0.460%,均值为0.336%;20批炒火麻仁饮片标准汤剂胡芦巴碱含量转移率为83.93%~98.72%,均值为91.11%。
2.3 超高效液相色谱法(UPLC)特征图谱研究
2.3.1 色谱条件与系统适用性试验 ACQUITY UPLC HSS T3 色谱柱(100 mm×2.1 mm,1.8 μm),流动相为乙腈(A)-水(B),梯度洗脱(0~6 min,2%~12%A;6~14 min,12%~40%A;14~16 min,40%~80%A;16~21 min,80%A;21~24 min,80%~100%A;24~26 min,100%A);流速为0.30 mL·min-1;检测波长为205 nm;柱温为35 ℃;进样量为1 μL。
2.3.2 对照品溶液的制备 分别取对照品α-亚麻酸和亚油酸适量,精密称定,加甲醇制成质量浓度为10 μg·mL-1的溶液;取鸟苷对照品适量,精密称定,加水制成质量浓度为20 μg·mL-1的溶液,作为对照品溶液。
2.3.3 供试品溶液的制备 取炒火麻仁饮片标准汤剂冻干粉,研细,取约0.1 g,精密称定,置具塞锥形瓶中,精密加入50%甲醇10 mL,密塞,称定质量,超声(500 W,40 kHz)30 min,取出,放冷,再称定质量,用50%甲醇补足减失的质量,摇匀,滤过,取续滤液,即得。
2.3.4 对照品指认 精密吸取2.3.2 项下对照品溶液和2.3.3 项下供试品溶液各1 µL,进样测定,记录色谱图,结果见图1。

图1 对照品及炒火麻仁饮片标准汤剂的UPLC图
结果表明,在炒火麻仁饮片标准汤剂特征图谱中,分别呈现与鸟苷(峰1)、α-亚麻酸(峰5)和亚油酸(峰6)保留时间一致的特征峰。
2.3.5 特征图谱方法学考察 精密度试验:取同一份炒火麻仁饮片标准汤剂供试品溶液,按照2.3.1项下色谱条件连续进样6次,记录色谱图,并以α-亚麻酸峰(峰5)为参照峰,计算各特征峰的相对保留时间和相对峰面积,结果其RSD 均小于2%,表明仪器精密度良好。
稳定性试验:取同一份炒火麻仁饮片标准汤剂供试品溶液,分别在0、2、4、8、10、12、16、20、24 h按2.3.1项下色谱条件进样测定,以α-亚麻酸峰(峰5)为参照峰,计算各特征峰的相对保留时间和相对峰面积,结果其RSD 均小于2%,表明供试品溶液在24 h内稳定。
重复性试验:取同一批炒火麻仁饮片标准汤剂冻干粉,按2.3.3 项下方法平行制备6 份供试品溶液,按照2.3.1 项下色谱条件进样测定,以α-亚麻酸峰(峰5)为参照峰,计算各特征峰的相对保留时间和相对峰面积,结果其RSD 均小于3%,表明该方法重复性良好。
2.4 特征图谱样品检测与分析
取20 批炒火麻仁饮片标准汤剂冻干粉,按2.3.3 项下方法制备供试品溶液,按2.3.1 项下色谱条件进样测定,记录色谱图。将20 批样品色谱图导入“中药色谱指纹图谱相似度评价系统”(2012 版)软件中进行色谱峰匹配及相似度分析,选择峰面积稳定的色谱峰作为共有峰,最终确定6 个共有峰。20 批炒火麻仁饮片标准汤剂的特征图谱见图2,对照特征图谱见图3,相似度结果见表3。结果显示,20 批炒火麻仁饮片标准汤剂特征图谱与对照特征图谱之间的相似度均在0.90 以上,表明各批次炒火麻仁饮片标准汤剂具有较好的一致性。

表3 20批炒火麻仁饮片标准汤剂特征图谱相似度结果

图2 20批炒火麻仁饮片标准汤剂UPLC特征图谱
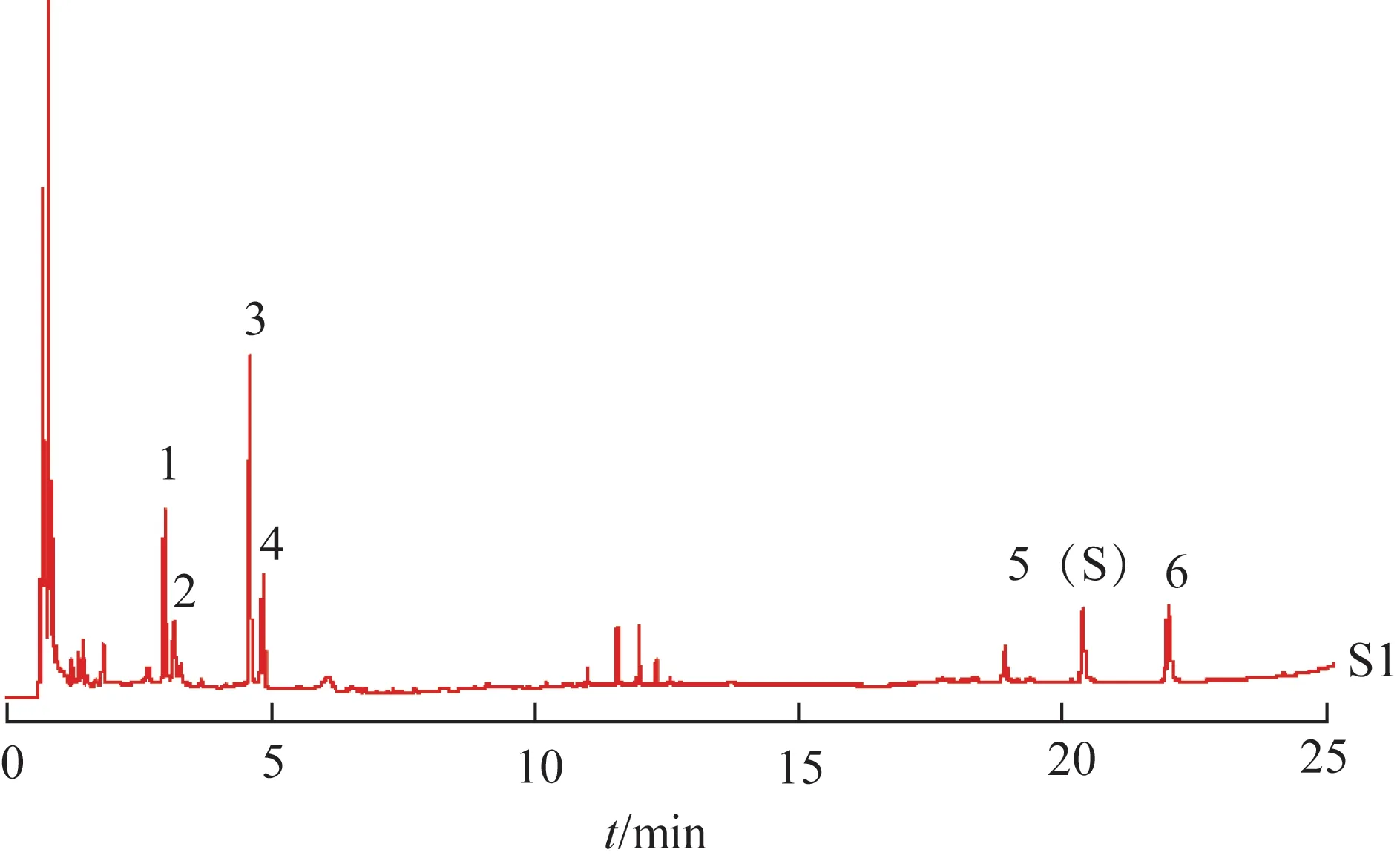
图3 炒火麻仁饮片标准汤剂对照特征图谱
3 讨论
3.1 标准汤剂制备工艺的合理性
本研究选取20 批合格的炒火麻仁饮片,参照《中药配方颗粒质量控制与标准制定技术要求》[9]和《医疗机构中药煎药室管理规范》[10]的煎煮方法,以水为提取溶剂,煎煮2 次,经固液分离、低温减压浓缩、冷冻干燥后制得样品。炒火麻仁以果实入药,为泻下药,不属于清热、滋补类药。所以炒火麻仁饮片标准汤剂一煎煮沸后再煎煮30 min,二煎时间适当缩短为25 min。本研究考察了过滤方式及不同目数筛网的过滤效果,水煎液直接过筛时,会残留较多油脂在筛网上。因此,通过先将水煎液油层吸取出来,再将剩余水煎液过筛的方式,可以尽量收集水煎液中的油脂。通过对200目和350目筛网的比较发现,两者滤液中均无明显可见固体杂质,为了提高过滤效率,选择了200 目筛网进行固液分离。同时通过比较不同浓缩温度下炒火麻仁饮片标准汤剂的密度、出膏率及浓缩液状态,发现在65 ℃减压条件下进行浓缩时,浓缩液可正常起泡蒸发,终密度为1.00~1.02 g·mL-1,为了保证标准汤剂质量的稳定性,最终采用65 ℃减压进行浓缩。
3.2 特征图谱方法的考察
通过二极管阵列检测器对炒火麻仁饮片标准汤剂中特征图谱色谱峰信号进行全波长(190~400 nm)扫描,再结合炒火麻仁饮片中油脂类成分,如亚油酸、α-亚麻酸等分子结构中仅含有非共轭双键,因此只有末端吸收,全波长扫描结果也显示,炒火麻仁饮片标准汤剂在205 nm 附近具有较强吸收,因此选择205 nm 作为特征图谱的检测波长。流动相考察了乙腈-水、乙腈-0.1%磷酸水溶液和甲醇-水3 种不同体系,标准汤剂供试品溶液在乙腈-0.1%磷酸水溶液和甲醇-水流动相体系中基线漂移较大,在乙腈-水体系中特征峰个数、响应值及基线平稳程度均较优,因此采用乙腈-水作为炒火麻仁饮片标准汤剂特征图谱的流动相体系。考察了30、35、40 ℃柱温的差异,结果在柱温35 ℃时火麻仁标准汤剂主要色谱峰的分离度更好。
供试品溶液制备方法时考察了无水乙醇、甲醇、50%乙醇水溶液和水4 种溶剂对炒火麻仁饮片标准汤剂的提取效率,结果50%甲醇提取的峰个数最多,提取效率最高;同时比较了超声提取和回流提取2种方式对提取效率的影响,结果2 种提取方式差异不大,从操作方便考虑,选择超声提取。
3.3 有效成分检测方法的建立
参考文献[11]中关于火麻仁饮片中胡芦巴碱的研究采用的是高效液相色谱法(HPLC),样品处理过程复杂,且对色谱柱的要求较高。考虑到色谱柱的通用性和普及性,本研究选用了C18反向键合色谱柱,简化了样品的处理过程,并且随着仪器的普及和检测器的多样化,UPLC 以其高效、快速、灵敏、节省溶剂等优点,在中药指纹图谱和有效成分的分析上占据明显优势[12]。本研究采用UPLC 技术,建立了炒火麻仁饮片和标准汤剂中胡芦巴碱的含量测定方法,该法简便、易行,可有效测定炒火麻仁饮片中胡芦巴碱的含量。
4 结论
20 批炒火麻仁饮片标准汤剂的特征图谱,通过拟合共确定了6 个特征峰,并通过对照品指认了鸟苷、α-亚麻酸和亚油酸3 个成分,将多批结果导入“中药色谱指纹图谱相似度评价系统”(2012 版)进行计算,结果20 批炒火麻仁饮片标准汤剂样品的特征图谱相似度均高于0.90,表明该方法简便准确、重复性好,可用于炒火麻仁饮片标准汤剂的质量控制。
本研究根据《中药配方颗粒质量控制与标准制定技术要求》[9]中相关规定,确定了炒火麻仁饮片标准汤剂制备的相应工艺参数,制备了20 批不同产地的炒火麻仁饮片标准汤剂,并从指标性成分的定量检测及其出膏率、转移率和特征图谱等方面进行了系统研究,建立的指标性成分的定量检测方法和特征图谱方法可同时用于炒火麻仁饮片和标准汤剂的检测,研究结果可为炒火麻仁饮片标准汤剂和配方颗粒的制备及质量控制研究提供参考,从而提高产品质量控制水平。
猜你喜欢
基层中医药(2022年5期)2022-10-24 01:27:40
中国药学药品知识仓库(2022年5期)2022-04-11 21:25:52
世界科学技术-中医药现代化(2021年5期)2021-11-05 06:55:18
中国民间疗法(2021年14期)2021-08-30 08:24:56
食品研究与开发(2021年14期)2021-01-18 06:06:59
中成药(2019年12期)2020-01-04 02:02:50
长寿(2019年7期)2019-07-25 11:36:26
中国中医药现代远程教育(2019年8期)2019-01-29 14:30:24
天然产物研究与开发(2018年2期)2018-04-04 02:01:14
中国中医药现代远程教育(2014年18期)2014-03-01 04:30:05
