
1例短链酰基辅酶A脱氢酶缺乏症的诊断分析*
2022-12-25 08:07于春宇姜盼盼孙智勇杨江涛辜清泉吴莉萍毛久忠
罕少疾病杂志 2022年12期
于春宇 姜盼盼 孙智勇,* 杨江涛 辜清泉 吴莉萍 毛久忠
1.朝阳市妇幼保健计划生育服务中心 (北京 122000)
2.深圳爱湾医学检验实验室 (广东 深圳 518000)
3.深圳罕见病代谢组学精准医学工程研究中心 (广东 深圳 518000)
短链酰基辅酶A脱氢酶缺乏症(short-chain acyl-CoA dehydrogenase deficiency,SCADD)是由于短链酰基辅酶A脱氢酶(short-chain acyl-CoA dehydrogenase,SCAD或ACADS)基因缺陷造成的一种脂肪酸氧化代谢障碍疾病[1]。近年来随着串联质谱新生儿疾病筛查的广泛开展,大部分无临床症状SCADD婴幼儿通过新生儿筛查被发现[2-3]。基因方面,ACADS突变检测是确诊SCADD的金标准。生化方面,血液中丁酰肉碱(butyrylcarnitine,C4)和尿液中乙基丙二酸(ethylmalonic acid,EMA)升高是本病的主要特点[3]。C4与EMA可通过新生儿筛查干血片氨基酸及酰基肉碱和尿有机酸谱检测,但尿中乙基丙二酸升高并非SCADD的特异性改变,也会出现在戊二酸血症II型和线粒体病种[4]。因此,通过血串联质谱筛查配合尿有机酸检测和基因测定可以对SCADD精准诊断。
1 对象与方法
1.1 病例对象先证者,女,二胎,孕37周剖腹产, 出生体重2800g,出生后母乳喂养,有时吐奶,大便正常,因黄疸和严重病毒性感染来院治疗。家族史:父母非近亲婚配,身体健康,同胞哥哥,9岁,身体健康;家族中无类似疾病病史,否认家族遗传性疾病病史。体格检查:患儿2个月6天时,体格检查显示精神反应好,可竖头片刻;体重5公斤,头围39.5厘米,身长60厘米;面色发黄,哭闹时皮肤发花,下肢及背部散在片状青斑;余项检查无明显异常。
1.2 检测方法
1.2.1 常规检测 新生儿筛查常规三项(PKU,CH,CAH),血常规,血生化,血氨及超声检测。
1.2.2 氨基酸及酰基肉碱谱检测 采集足跟血制备的干血斑滤纸片,送深圳爱湾医学检验实验室通过串联质谱法(LC-MS/MS)进行氨基酸及酰基肉碱谱检测,并对可疑患者进行复查确认。
1.2.3 尿有机酸检测 可疑患儿留取新鲜尿液制备尿滤纸片,送深圳爱湾医学检验实验室通过气相色谱-质谱(GC-MS)检测尿液中有机酸水平,并进行复查确认。
1.2.4 基因检测 经医学伦理审核以及患儿法定监护人知情同意,采集可疑患儿及父母和同胞哥哥外周血2mL,运用外显子捕获测序技术对先症者及家庭成员的进行基因检测,按照ACMG遗传变异解读规范进行筛选和注释可疑的突变,寻求证据,得出突变的致病性结论。
1.3 治疗与随访患儿至北京大学第一医院门诊进行复诊,行药物及饮食干预,间隔2~3月门诊随访。
2 结 果
2.1 可疑患儿常规检测医院行血常规检测显示中性粒细胞数、中性粒细胞比率、红细胞计数和红细胞压积均下降,淋巴细胞数、淋巴细胞比率、单核细胞数、单核细胞比率和红细胞体积分布宽均升高,见表1。血生化检测显示血清总蛋白、球蛋白和血钠偏低,γ-谷氨酰基转肽酶、白/球比例、总胆红素、直接胆红素、直接胆红素、α-羟丁酸脱氢酶、肌酸激酶同工酶和血钾均偏高,见表2。血氨检测38.00(9~47μmol/L),显示正常。新生儿疾病筛查常规三项(PKU、CH、CAH)结果正常,超声心动图显示房间隔缺损。
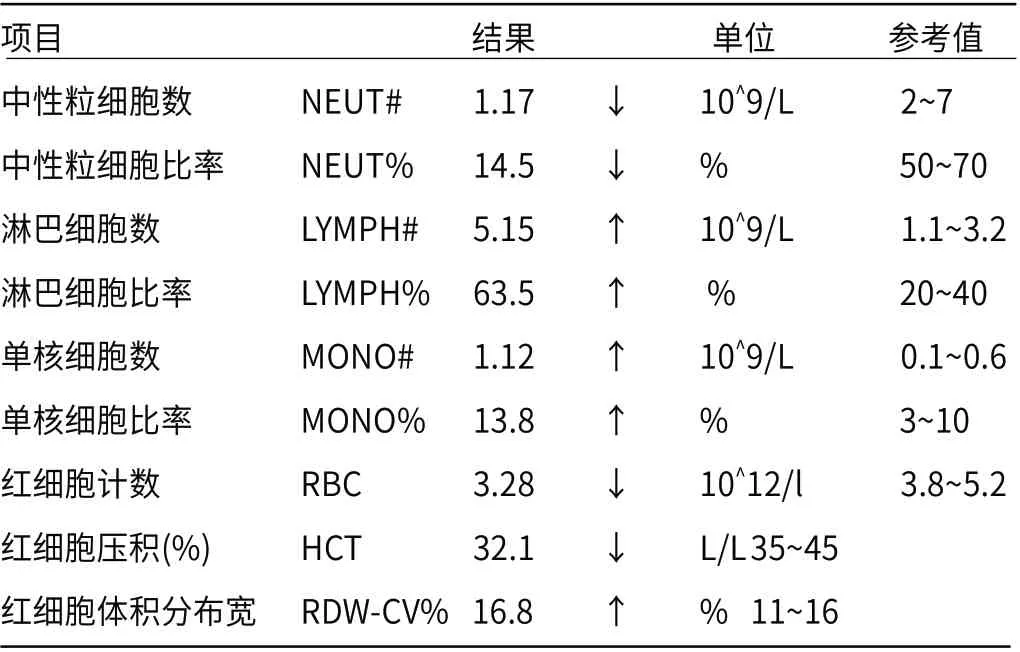
表1 血常规报告异常指标

表2 血生化检测异常指标
2.4 基因检测在患儿ACADS基因发现c.164C>T/c.431C>T复合杂合突变,其中c.164C>T为已知致病突变,c.431C>T为意义未明突变。在父亲ACADS基因检测到c.164C>T杂合突变以及UGT1A1基因c.211G>A杂合突变(Crigler- Najjar综合征),父亲是c.164C>T突变携带者。在母亲和哥哥ACADS基因检测到c.431C>T杂合突变,是c.431C>T突变携带者,见表5。至此,家系遗传模式确定,见图1,c.431C>T突变等级也从意义未明升级为疑似致病突变,父母双方均为ACADS基因致病突变携带者,导致患儿在ACADS基因形成复合杂合突变,确诊为短链酰基辅酶A脱氢酶缺乏症。

图1 遗传模式。图2 SCADD诊断流程。
2.2 氨基酸及酰基肉碱谱检测血串联测定结果显示丁酰肉碱(C4)及丁酰肉碱/乙酰肉碱(C4/C2),丁酰肉碱/丙酰肉碱(C4/C3)比值升高,见表3;提示可能为短链酰基辅酶A脱氢酶缺乏症,异丁酰辅酶A脱氢酶缺乏症或乙基丙二酸尿症,建议尿液有机酸分析,必要时基因分析进一步病因检查。
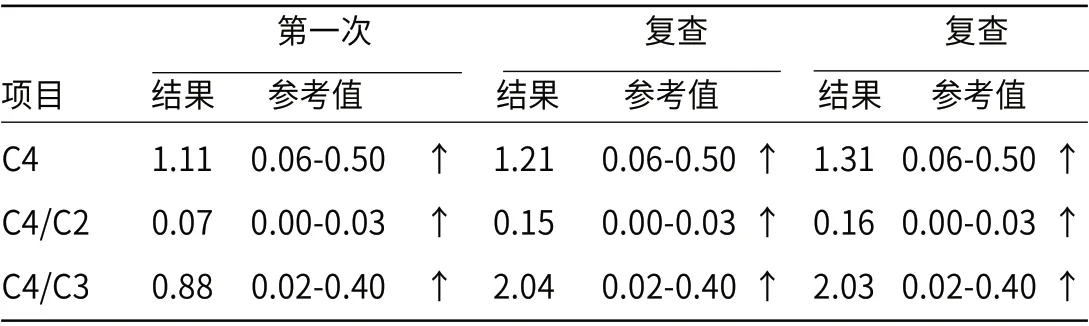
表3 酰基肉碱谱检测
2.3 尿有机酸检测串联质谱结果显示乙基丙二酸-2特异性指标倍数偏高,见表4。考虑短链酰基辅酶A脱氢酶缺乏症或乙基丙二酸脑病;建议联系家长进行基因分析,进一步确认病因。

表4 尿有机酸谱检测
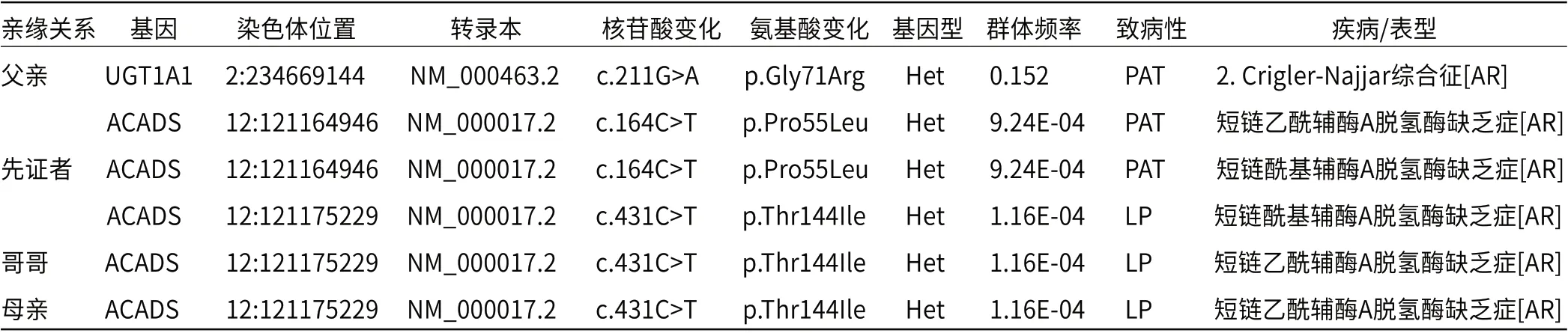
表5 家系基因检测结果
2.5 治疗与随访患儿予以左卡尼汀(0.5g/d)、伊可新(1粒/d)、复合维生素B(1粒/d)、维生素B2(10mg/d)、整肠生(0.25g/d)等对症支持治疗治疗,正常喂养。母亲予以爱乐维或金施尔康(1粒/d)和左卡尼汀(0.5g/d)口服。3个月后复诊,患儿智力、运动正常,肌张力正常,但有营养不良、贫血、缺铁等情况,另外给患儿和母亲予以蛋白琥珀酸铁补充用于改善相应,嘱其定期对方复查。至此,使用贯穿组学技术对患儿实现精准诊疗并获得到及时干预和良好预后,疾病诊断流程,见图2。
3 讨 论
短链酰基辅酶A脱氢酶缺乏症(SCADD)临床症状缺乏特异性,从婴儿期的致命失代偿性代谢性酸中毒到无症状个体均存在,严重程度不一。目前已报道患者中,有症状和无症状病例各占50%左右。对于有症状患者,不同发病年龄会出现不同的临床表现。新生儿期SCADD以肌张力低下和黄疸异常为主要共同特征;早发型患者的临床症状比较复杂,包括发育迟滞、肌张力下降、癫痫、低血糖、喂养困难、呕吐等各种症状;晚发型SCADD患者通常出现在儿童期或青少年期,大多伴有神经系统症状,包括发育迟缓、癫痫发作、畸形和自闭症等。随访中发现,与无症状患者相比,有症状患者的预后情况较差,其中约40%表现出语言、运动技能迟缓和智力发育障碍[5-7]。本例患儿出生临床表现正常,但在遗传代谢性疾病筛查过程中发现酰基肉碱谱(C4、C4/C2、C4/C3)指标异常,临床怀疑存在遗传代谢病的可能性,随即对其进行病因诊断。
在我国仅有部分地区将此病列入新生儿疾病常规筛查项目,筛查指标没有完全涵盖血酰基肉碱和尿有机酸及基因测定。新生儿遗传代谢病筛查检测到C4 ,C4/C3,C4/C2指标异常,这3个指标异常会提示至少3种疾病,分别为异丁酰辅酶A脱氢酶缺乏症,乙基丙二酸脑病,短链酰基辅酶A脱氢酶缺乏症[16]。通过进一步行尿液有机酸谱测定乙基丙二酸指标异常才能排除异丁酰辅酶A脱氢酶缺乏症。至此,对患者再行ACADS 基因测定,完成短链酰基辅酶A脱氢酶缺乏症精准诊断。但美国也曾报道过 1例SCADD 患儿血C4和尿乙基丙二酸含量均在正常范围内,通过基因分析检测到c.625G>A/c.511 C>T复合杂合突变,进而确诊为SCADD[8]。如果在新生儿筛查阶段进行氨基酸及酰基肉碱谱和基因联合筛查,可以降低漏筛,漏诊的情况。
短链酰基辅酶A脱氢酶缺乏症(SCADD)治疗尚无统一共识,目前文献报道中大部分病例治疗的主要通过改善临床症状,低脂饮食,可适当补充肉碱或维生素B2(核黄素),避免长时间禁食[9-12]。急性发作期,可静脉给予10%葡萄糖溶液,速率为8~10mg/(kg·min)或者口服葡萄糖液一直分解代谢[13]。对于补充肉碱治疗一直存在争议,通过摄入左旋肉碱增加C4代谢,减少尿EMA排出的可行性及有效性仍需被证实[14]。本例患儿由于处在新生儿哺乳期,因此在治疗中不仅针对患儿采取了补充肉碱、核黄素等方式,还给予母亲肉碱等补充用以改善患儿的症状,取得了较好的效果。
猜你喜欢
中老年保健(2022年4期)2022-08-22
农业工程学报(2021年3期)2021-04-15
茶叶(2020年1期)2020-05-30
山东农业科学(2019年9期)2019-12-09
华声文萃(2019年4期)2019-09-10
文萃报·周二版(2019年10期)2019-09-10
无机化学学报(2019年2期)2019-02-27
食品界(2017年4期)2017-05-17
家庭百事通·健康一点通(2017年3期)2017-03-22
武术研究(2011年10期)2011-08-15
