
煤对甲烷及氘代甲烷吸附性能研究
2022-12-24 07:00岳基伟孙永鑫邵亨威吕振威李怀宾
煤炭科学技术 2022年11期
岳基伟,李 宏,孙永鑫,邵亨威,吕振威,李怀宾
(1.安徽理工大学 安全科学与工程学院,安徽 淮南 232001;2.河南工程学院 安全学院,河南 郑州 451191;3.河南理工大学 安全科学与工程学院,河南 焦作 454000)
0 引 言
煤对甲烷的吸附特性可以用来表征煤体吸附甲烷的能力,研究煤对甲烷的吸附特性对于矿井瓦斯灾害防治具有重要的意义[1-2]。水分是影响煤对甲烷吸附、解吸的因素之一[3-4],水力化措施是煤层引入外加水分的主要途径之一,在煤炭开采领域水力化措施对防突、防冲、防尘具有重要的作用[5]。水力化措施实施过程中(如煤层注水、水力压裂等)主要以驱替游离瓦斯为主,实施后的水分会发生渗吸效应,造成吸附态瓦斯被置换为游离态瓦斯[6]和含瓦斯煤体孔隙被有效润湿,渗吸平衡后,水吸附孔隙表面形成水膜,阻碍瓦斯扩散,造成水对微孔隙瓦斯的封堵效应[7]。因此,弄清水分在煤-甲烷-水三相体系中的分布对于揭示水分在含瓦斯煤中的运移机制具有重要的意义。
测试水分分布的方法有干燥法,电阻率法,红外线法及NMR测试法等。干燥法测试多孔介质中含水率精度较高,但干燥法无法实现煤-甲烷-水三相体系中水分含量的实时测定[8]。YUE等[9]采用电阻率法测试了含瓦斯煤自发渗吸过程水分的运移距离,电阻率法可以从宏观角度测试水分的运移,无法定量获得水分在孔隙内分布。红外线的穿透能力很弱,无法穿透钢制品[10]。NMR测试技术作为一种无损检测技术,被广泛应用于研究微观尺度上流体在多孔介质中的流动及空间分布特性[11-15]。然而,NMR测试技术以氢原子核的响应为基础[16]。NMR测试技术在检测不含瓦斯煤中水分的分布时具有独特的优势,因为水分子中氢原子核具有较大的磁矩及较强的信号。当NMR测试技术被应用于水与瓦斯两相运移机理研究时,如果采用重水代替水时,可获得吸附态甲烷、游离态甲烷的变化规律,不能获得水分的分布。因此,亟待寻找一种测试方法能够测试煤-甲烷-水三相体系中水分的分布。
基于此,笔者提出采用一种吸附性气体-氘代甲烷(CD4)代替甲烷进行研究。研究表明,煤对CO2、CH4及N2的吸附能力依次减小[17]。然而,煤-CO2-水分三相共存时,CO2与水会产生碳酸,从而对煤体产生溶蚀效应。CD4是CH4中的氢原子被氘原子取代而形成的物质,CD4分子是否可以代替CH4作为吸附质研究水与瓦斯两相共存时水分的分布及运移规律的关键是弄清煤对CH4及CD4吸附性能的差异。
笔者采用试验的方法及分子动力学模拟法,研究煤对CH4及CD4的吸附性能,研究结果为采用NMR测试煤-CD4-水三相体系中水分的分布奠定理论基础。
1 煤对甲烷及氘代甲烷吸附性能试验
试验煤样取自河南焦作古汉山矿,对工作面取回的新鲜煤样进行破碎筛选,筛选出粒径为60~80目(0.250~0.180 mm)煤样,按照国家标准GB/T 212—2008《煤的工业分析方法》进行煤样的工业分析,煤样的水分Mad为2.58%,灰分Aad为16.66%,挥发分Vdaf为8.96%。采用H-Sorb 2600T高低温高低压吸附解吸扩散仪,按照GB/T 19560—2008《煤的高压等温吸附试验方法》测试不同温度、不同瓦斯压力条件下煤样的瓦斯吸附量,测试结果如图1所示。

图1 不同温度及不同瓦斯压力下煤对CH4及CD4的吸附量Fig.1 Adsorption capacity of coal to CH4 and CD4 under different temperature and gas pressures
由图1可知,在0~3 MPa内,煤对CH4及CD4的吸附量随着吸附平衡压力的增加而增加,增加的梯度逐渐减小,主要原因是平衡压力的增加,导致CH4及CD4分子撞击煤孔隙表面的机率增加,CH4及CD4分子在煤孔隙表面的撞击次数、个数、撞击面积增加,因此气体分子在煤孔隙表面的吸附相密度增加,吸附相密度的逐渐增加导致煤对瓦斯的吸附位逐渐减小,因此煤对瓦斯的吸附量随着吸附平衡的压力的增加而增加,增加的梯度逐渐减小。煤对瓦斯的吸附量与吸附平衡压力满足Langmuir的吸附模型如式(1)所示:
Q=abp/(1+bp)
(1)
式中:p为吸附平衡压力,MPa;Q为吸附量,mL/g;a和b为吸附常数。
拟合参数见表1。在相同的温度条件下,煤对CH4及CD4的吸附量基本相同,且吸附常数a值差别不大。CH4变为CD4是氘取代了氢,化学性质没有发生改变,键长键角均相同。

表1 Langmuir吸附模型的拟合参数
2 分子动力学模拟方法及吸附构型
2.1 分子模拟方法
采用Materials Studio分子模拟软件构建并优化无烟煤大分子结构模型,运用分子动力学方法(MD)、统计学系中的正则系综(NVT)对CH4分子及CD4分子进行不同温度、不同压力条件下吸附量、吸附能的模拟及理论计算[18]。CH4分子中的C—H键的键长和CD4分子中的C—D键的键长相等,均为0.108 4 nm,H—C—H键角均为109.5°[19]。CH4分子及CD4分子结构如图2所示。
建立表层嵌贴FRP板条-混凝土界面黏结-滑移模型的方法······························张智梅 张振凯 熊 浩 王 卓 陈 刚 (2,304)
图2 CH4及CD4分子结构Fig.2 Molecular structure of methane and deuterated methane
模拟使用的是COMPASSⅡ力场,在进行分子动力学计算时,采用周期性边界条件对系统进行优化处理,使分子模拟系统的性质接近于真实系统[20]。在周期性边界条件下,分子被封闭在一个盒子中作为模拟单元,假定其周围包围着无穷多个与模拟单元内部的粒子数量排列方式、速度完全等价的盒子。当从盒子中移出某一粒子后,一定会有另一粒子进入,保持盒子中的总粒子数不变,使得结构空间无限延伸,克服了分子数目的限制,采用截断半径法计算远程分子间的相互作用力。
2.2 无烟煤微晶吸附CH4及CD4构型
使用分子动力学Ms. Sorption和分子力学Ms. Forcite模块对CH4及CD4分子在无烟煤中的吸附性能进行模拟计算。分子结构采用WENDER等[21]提出的无烟煤的大分子结构模型,众多学者采用X射线衍射(XRD)方法对煤微晶结构进行了研究,并提出煤的大分子结构是一种由非晶体向晶体过渡的微晶结构。因此,先将构建无烟煤超晶胞分子模型放入到周期性边界的晶体盒子里,盒子里设置空间群1P1,晶格的长宽高分别设置为a’=2 nm,b’=1.2 nm,c’=5 nm;角度分别为(α=90°,β=90°,γ=60°)。为给微晶模型提供足够的吸附/扩散空间,使用Cleave Surface命令沿着(0,0,-1)平面将晶格割开,留设真空层以模拟煤分子结构中的孔隙。甲烷在无烟煤微晶中的吸附构型如图3所示。

图3 无烟煤微晶吸附CH4及CD4的构型Fig.3 CH4 and CD4adsorption configuration in anthracite microcrystalline
无烟煤微晶模型的等温吸附和吸附构型均基于GCMC原理,在Sorption模块下进行。等温吸附采用Adsorption Isotherm任务项模拟,总步长设定为2×107步,模拟的压力范围从0.01~7.00 MPa,模拟的温度为(253.15、273.15、293.15、313.15 K)。模拟中采用CompassⅡ力场,体系中的电荷由力场自动分配,静电力采用Eward计算,范德华力采用Atom Based;采样方法选用Metropolis准则进行,计算精度为Fine。等温吸附构型采用Locate任务项进行计算,其他参数与Adsorption isotherm任务项中设置一致。
3 煤对CH4及CD4吸附性能的分子动力学模拟
CH4及CD4在煤中的吸附是分子之间相互作用力的结果,其中吸附态占据大多数,少部分以游离态存在。采用分子模拟的方法从吸附量、吸附热2个方面研究煤对CH4及CD4吸附性能的差异性。不同温度、不同压力条件下无烟煤分子吸附CH4及CD4情况如图4所示。
将图4中不同温度、不同压力条件下的吸附分子数代入式(2)[22],可以得到CH4及CD4在无烟煤单个分子层上的吸附量。
N=1 000Nam/(NAMs)
(2)
式中:N为甲烷的吸附量,mmol/g;Nam为模拟计算的吸附分子数,个;NA为阿伏伽德罗常数,6.02×1023;Ms为单个晶胞的分子质量,Ms=9.484 8×10-21g。
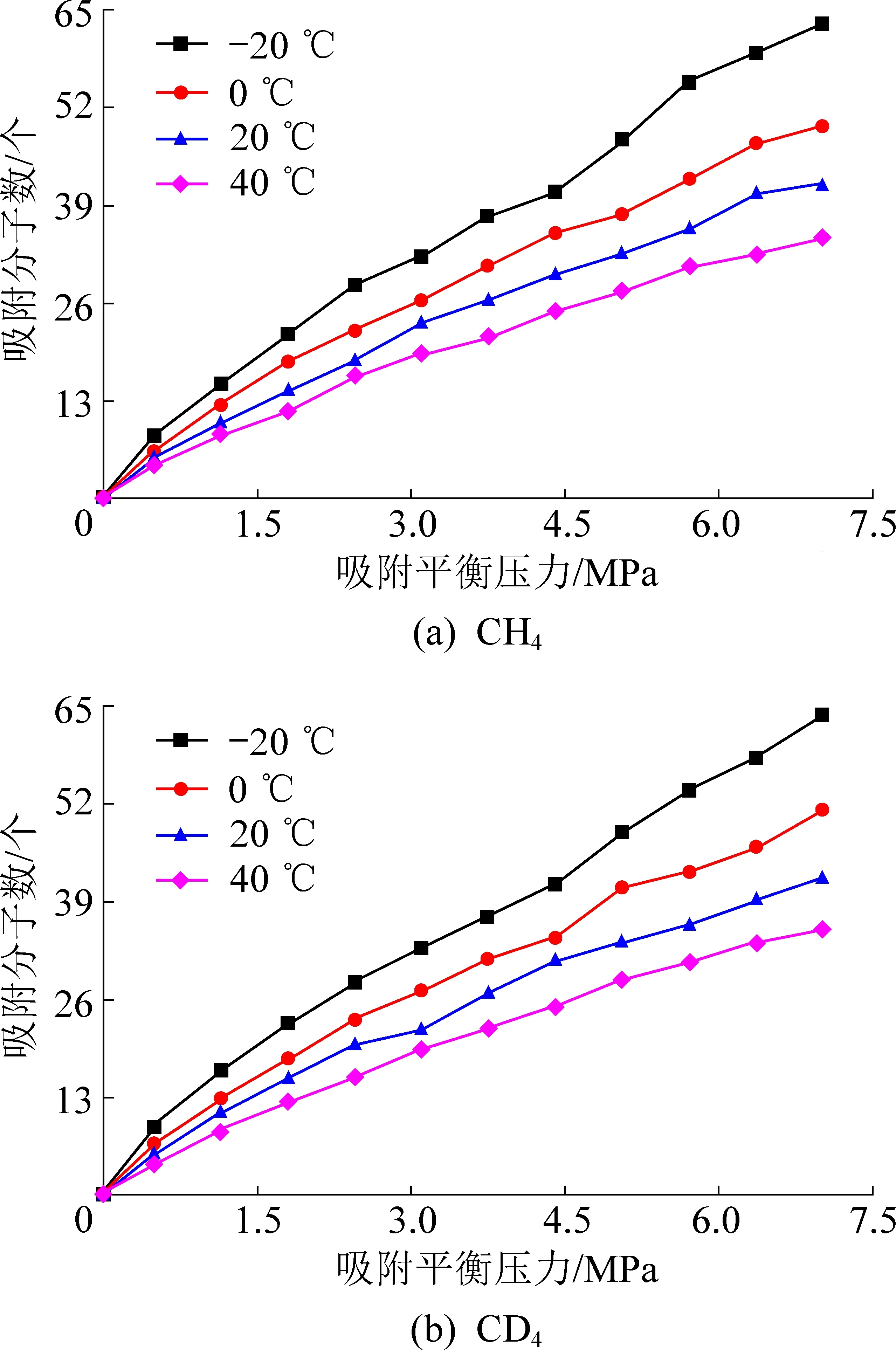
图4 不同温度、不同平衡压力条件下煤对CH4及CD4 吸附情况Fig.4 Adsorption of CH4 and CD4 by coal under different temperatures and equilibrium pressures
3.1 不同压力条件下煤对CH4及CD4吸附量模拟
同一温度,不同压力条件下煤对CH4及CD4吸附量如图5所示。

图5 不同压力条件下煤对CH4及CD4的吸附量Fig.5 Adsorption capacity of coal for CH4 and CD4 under different pressure conditions
由图5a—图5d可知,煤对CH4及CD4的吸附量随着吸附平衡压力的增加而增加,吸附量与吸附平衡压力满足Langmuir的函数关系,拟合参数见表2。
3.2 不同温度条件下煤对CH4及CD4的吸附量模拟
温度为-20 ℃、0 ℃、20 ℃及40 ℃条件下煤对CH4及CD4的吸附量如图6所示。由图6可知,煤对CH4及CD4的吸附量均随着温度的升高而减小,低温环境有助于CH4及CD4分子在煤微晶结构中被吸附,降温可以降低游离CH4及CD4分子的动能,CH4及CD4分子更易被范德华力所“捕获”;温度越高,CH4及CD4分子热运动越剧烈,平均动能增加,脱离范德华力束缚的概率增大,导致相同压力下煤对CH4及CD4的吸附量降低。

表2 式(1)拟合参数

图6 不同温度条件下煤对CH4及CD4的吸附量Fig.6 Adsorption capacity of coal for CH4 and CD4 at different temperatures
4 煤对CH4及CD4的等量吸附热
等量吸附热是区分化学吸附和物理吸附的重要热力学参数,当吸附热大于42 kJ/mol时为化学吸附,吸附热小于42 kJ/mol时为物理吸附;同时吸附热还是衡量CH4在无烟煤结构模型中吸附强弱的指标。分子动力学中模拟等量吸附热是根据吸附等温线中相同吸附量对应的温度和压力,采用克劳修斯-克拉珀龙(Clausius-Clapeyron)方程[23](式(3))计算得到,计算结果如图7所示。
qst=RT2d(lnp)/dT
(3)
式中:qst为等量吸附热,kJ/mol;R为理想气体常数,8.314 J/(molK);T为试验温度,K。
由图7a可知,等量吸附热随着吸附平衡压力的增加而减小,在等吸附量的条件下,温度越高,吸附平衡压力越大,吸附过程越不易进行,放出的热量越小。因此,等量吸附热随着吸附平衡压力的增加而减小。因此,等量吸附热随着吸附平衡压力的增加而减小。
CH4及CD4的等量吸附热随着温度降低而逐渐增加。因为温度降低使CH4及CD4分子平均动能减小,受范德华力的作用,被煤微晶捕获的CH4及CD4分子增多,同时降温更利于促进CH4及CD4吸附,放出的热量增加,导致等量吸附热整体呈现出增加的趋势。
采用式(4)对图7中的数据进行拟合,拟合参数见表3,即可获得等量吸附热与吸附平衡压力的关系。由式(4)及表3可知,对于CH4而言,当试验温度为-20、0、20、40 ℃时,等量吸附热为10.462 33~12.247 27 kJ/mol,10.121 33~11.688 50 kJ/mol,9.830 76~11.115 40 kJ/mol,9.492 35~10.575 40 kJ/mol;对于CD4而言,当试验温度为-20、0、20、40 ℃时,等量吸附热为10.547 00 ~12.434 00 kJ/mol,10.040 00~11.195 10 kJ/mol,9.892 30~10.956 60 kJ/mol,9.357 00~10.819 20 kJ/mol。在同一温度条件下,煤对CH4及CD4的等量吸附热差别不大。

图7 煤对CH4及CD4的等量吸附热Fig.7 Isosteric adsorption heat of coal for CH4 and CD4

表3 最大吸附热及最小吸附热拟合参数
当吸附热大于42 kJ/mol时为化学吸附,吸附热小于42 kJ/mol时为物理吸附[24],由此可知煤对CH4及CD4的吸附均为物理吸附。
qst=d+fegp
(4)
式中:qst为等量吸附热, kJ/mol;d,f,g为拟合参数;p为吸附平衡压力,MPa。
5 结 论
1)煤对甲烷及氘代甲烷的吸附量差别不大,且吸附量与吸附平衡压力满足Langmuir的含数关系。
2)煤对甲烷及氘代甲烷的等量吸附热随着吸附平衡压力的增加而减小,等量吸附热与瓦斯压力满足指数的函数关系。
3)煤对CH4及CD4吸附的等量吸附热随着温度的减小而增加,且等量吸附热均存在极限值、均为物理吸附。
4)研究结果为采用核磁共振定量研究煤-甲烷-水分三相体系中水分的分布规律奠定理论基础。
猜你喜欢
新高考·高三数学(2022年3期)2022-04-28
矿山安全信息(2021年22期)2021-12-25
矿山安全信息(2021年17期)2021-12-25
陶瓷学报(2021年4期)2021-10-14
建材发展导向(2021年13期)2021-07-28
建材发展导向(2021年11期)2021-07-28
陶瓷学报(2021年1期)2021-04-13
昆钢科技(2020年5期)2021-01-04
数学大王·低年级(2017年11期)2017-12-05
中学生数理化·中考版(2016年7期)2016-12-07
