
ITPA基因相关发育性癫痫性脑病35型患儿的表型特点
2022-12-09 10:20:02王嘉欣万林李志超孙于林王静石秀玉杨光
癫痫与神经电生理学杂志 2022年5期
王嘉欣,万林,李志超,孙于林,王静,石秀玉,杨光
发育性癫痫性脑病(developmental and epileptic encephalopathy,DEE)35型是一种罕见的神经退行性疾病,因三磷酸肌苷焦磷酸酶(inosine triphosphate pyrophosphatase,ITPA)基因突变导致,临床主要表现为全面发育迟缓、癫痫发作、小头畸形以及特征性的颅脑影像学改变,预后差[1]。本文报道解放军总医院第一医学中心儿科确诊的1例DEE 35型病例的临床特点和诊治过程,并结合国内外文献总结DEE 35型的基因型和表型特征,进一步阐明及加深对DEE 35型表型的认识。
1 临床资料
患儿,女,2月龄,出生后25 d时出现无诱因抽搐发作,表现为双眼凝视、四肢强直阵挛,每日发作数次,就诊于我院门诊,视频脑电图(V-EEG)示发作间期双侧中颞区、左侧枕区痫样放电(图1),诊断为癫痫(全面强直阵挛发作),给予左乙拉西坦及托吡酯口服,并逐步调整剂量,发作有所减少。患儿系第二胎第二产,足月顺产,出生评分不详,出生后因“呻吟”于当地医院新生儿科住院治疗,期间查颅脑MRI未见明显异常。出生后2 d时出现黄疸伴消退延迟,约出生后1个月渐消退。父母非近亲结婚,体健。有一姐姐因“不明原因抽搐”于9月龄时去世,具体发作形式及病因不详。查体(2月龄):神志清,反应可,无特殊面容,周身皮肤无色素脱失斑及皮疹,四肢肌力、肌张力正常,生理反射正常引出,病理反射未引出。血尿代谢筛查未见明显异常。

注:脑电图示双侧中颞区、左侧枕区痫样放电。图1 患儿发作间期脑电图
遗传学检测:患儿家属签署知情同意书后,采集患儿及父母亲外周血行Trio全外显子组测序,结果显示,患儿ITPA基因存在复合杂合变异,一处为剪接变异,c.124+1G>A(母源),一处为无义变异,c.304C>T(p.Gln102*)(父源),根据美国医学遗传学与基因组学学会(ACMG)制定的遗传变异分类标准与指南分析均为疑似致病性(PVS1+PM2和PVS1+PM2),与DEE 35型相关(图2)。

注:A为患儿家系图,B为测序峰图(患儿及母亲ITPA基因存在c.124+1G>A剪接变异,父亲为野生型),C为测序峰图(患儿及父亲ITPA基因存在c.304C>T无义变异,母亲为野生型)。图2 患儿家系图以及患儿与父母亲的基因测序图
治疗及随访:根据患儿临床表现、脑电图、颅脑MRI及遗传学检测结果,明确诊断为发育性癫痫性脑病35型,予左乙拉西坦及托吡酯抗癫痫治疗,患儿发作频次减少。7月龄随访:平均每天发作1次,复查脑电图示双侧前中颞区痫样放电。12月龄随访:复查颅脑MRI示脑萎缩改变,胼胝体略细小(图3)。2岁时随访:出现双侧玻璃体浑浊,于当地医院眼科就诊诊断为先天性白内障,未进一步治疗。2岁零6月龄末次随访:口服左乙拉西坦1.5 mL,2次/d,托吡酯25 mg, 2次/d,无癫痫发作近1年;追声追物一般,可被逗笑,抬头不稳,不会爬、坐、扶站及走,不会叫“爸爸、妈妈”,可无意识发出“a、ma”音。
2 文献复习
以中文“ITPA基因”“癫痫”,英文“ITPAgene”“epilepsy”为关键词,在PubMed、NCBI、中国知网及万方数据知识服务平台等数据库中进行检索,共检索到英文文献14篇,中文文献1篇。截至2022年1月,包括本病例在内,国内外共报道14例ITPA基因相关癫痫患者,其中男8例,女6例。其中2例为ITPA基因复合杂合变异,12例为纯合子变异(其中10例患者父母有血缘关系)。多数患儿于婴儿期发病,几乎所有的患者都在婴儿期或幼儿期死亡(14例先证者中有10例在3岁前死亡,11例在5岁前死亡)。临床表型方面,除国内2例肌张力正常外,所有病例均表现为全面发育迟缓、癫痫发作、小头畸形及肌张力减低,并伴有特征性的颅脑影像学改变[胼胝体薄、脑萎缩和(或)髓鞘发育迟缓及内囊后肢白质改变],Kevelam等[17]在文献中报道了本病典型影像学表现(图4)。白内障和扩张性心肌病是其他特征性表现(8例患者有白内障,3例有心肌病,另1例显示左心室轻度扩张)[1-2]。治疗方面,国外所报道的12例患者均表现为难治性癫痫,几乎所有的患者都在早期死亡。国内除本例外,目前报道1例,经丙戊酸钠、奥卡西平、左乙拉西坦及生酮饮食等治疗,仍有点头发作(发作频率减少50%以上)。本例患儿经左乙拉西坦联合托吡酯治疗,随访至今,已无发作近1年。14例患儿的详细资料见表1。

注:颅脑MRI示脑萎缩改变,胼胝体略细小。图3 患儿12月龄时的头颅MRI

注:A~D为患者2,56月龄,E~H为患者4,54月龄,A~H显示髓鞘形成延迟,并有轻度的脑萎缩。C、G箭头示内囊后肢锥体束T2高信号,B、F箭头示大脑脚和大脑脚内锥体束的受累,E箭头示在延髓锥体、小脑下脚和小脑深部白质信号异常。图4 Kevelam 等报道的患者的头颅MRI
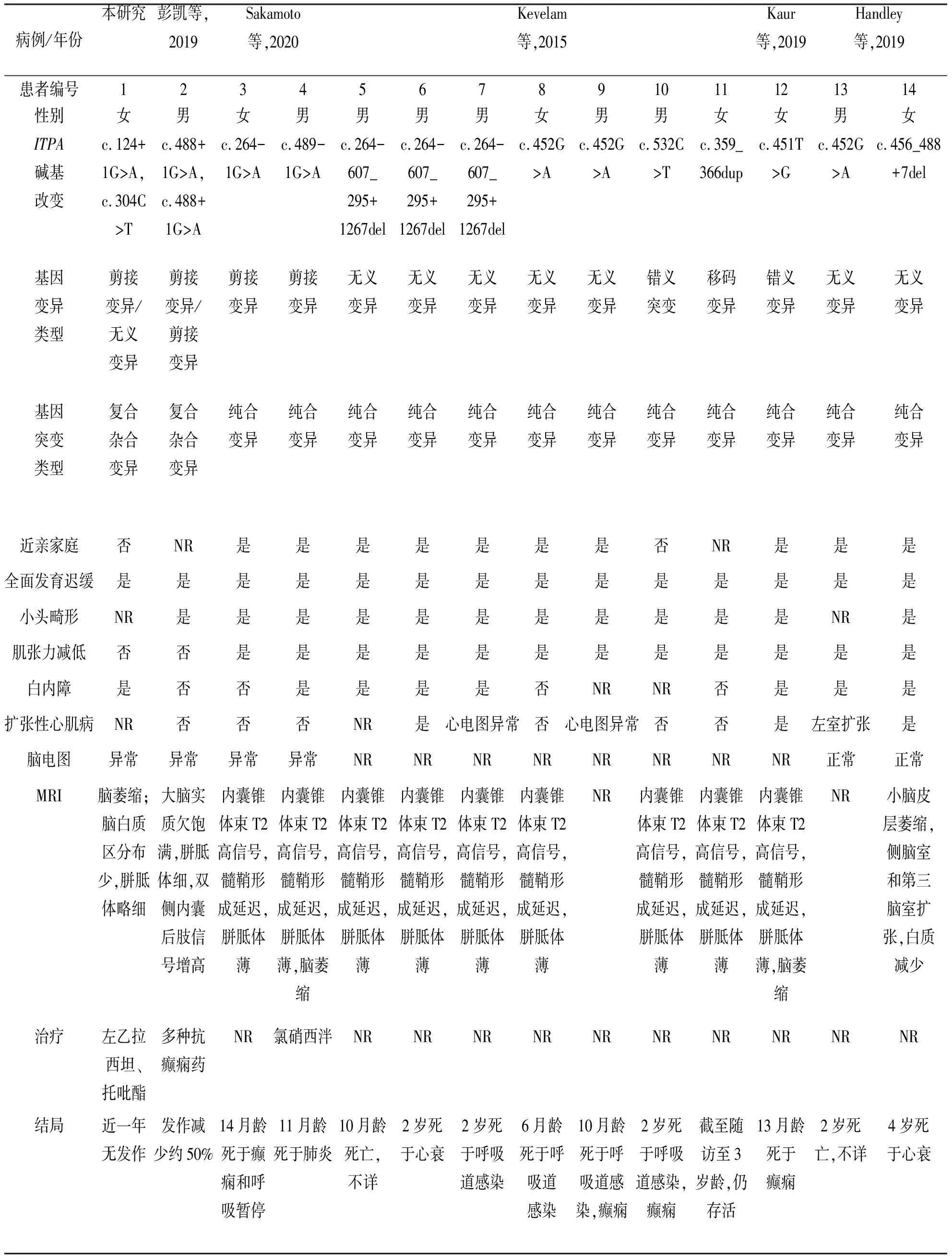
表1 既往文献报道的DEE 35型的基因型和表型特点
3 讨论
DEE是指癫痫患者出现的脑病与病因及癫痫活动均相关,主要由遗传原因导致,预后一般较差[3-4]。其中,DEE 35型是由编码三磷酸肌苷焦磷酸酶的ITPA基因变异所致的常染色体隐性遗传性疾病,主要表现为全面发育迟缓、癫痫发作、进行性小头畸形、肌张力减退、胼胝体变薄、脑萎缩、髓鞘形成延迟及内囊后肢白质改变等。部分患儿可有白内障和扩张性心肌病表现,其确诊依赖基因检测。
ITPA基因位于20号染色体的短臂上,编码三磷酸肌苷焦磷酸酶[5]。三磷酸肌苷焦磷酸酶属于HAM1 NTPase家族,是一种细胞内酶,定位于细胞质,在人体多组织中表达,是一种同源二聚体蛋白,每个单体由194个氨基酸残基组成[2,5-10]。ITPase通过将非规范的嘌呤核苷酸肌苷三磷酸和脱氧肌苷三磷酸水解为单磷酸核苷酸和二磷酸,水解产物进一步转化后与RNA和DNA结合,将非规范嘌呤排除在RNA和DNA前体库中,避免染色体损伤[2-3,5,11-12]。既往研究中,ITPA基因的某些变异被报道与免疫抑制药物硫唑嘌呤和6-巯基嘌呤的不良反应以及利巴韦林引起的溶血性贫血有关[13-16]。2015年,kevelam等[17]首次报道了ITPA基因变异与癫痫相关。目前认为ITPA基因变异时,可能通过影响三磷酸肌苷焦磷酸酶与非规范嘌呤结合,使非规范嘌呤过度累积,掺入RNA和DNA中而致病[3,6,12,18-20]。
复习国内外文献,包括本病例在内共有14例ITPA基因相关癫痫患者,其中2例为复合杂合变异,12例为纯合子变异。多数患儿于婴儿期发病,表现为多种形式的癫痫发作,除本例外均为药物难治性癫痫。除国内2例肌张力正常外,其余病例均表现为全面性发育迟缓、小头畸形、肌张力减低,并伴有特征性的颅脑影像学改变,包括胼胝体薄、脑萎缩和(或)髓鞘发育迟缓及内囊后肢白质改变等[1-3,17]。几乎所有的患者都在婴儿期或幼儿期死亡(14例先证者中有10例在3岁前死亡,11例在5岁前死亡)。
综上所述,ITPA基因变异导致的DEE 35型起病早,癫痫难以控制,预后较差。本例患儿发现的ITPA基因变异位点未见文献报道,为新发现的变异,增加了ITPA基因变异所致DEE 35型的表型和基因谱。
猜你喜欢
食品与发酵工业(2024年4期)2024-03-01 12:56:12
保健与生活(2022年9期)2022-05-06 22:50:00
科学生活(2019年7期)2020-01-01 08:28:02
养殖与饲料(2019年10期)2019-02-25 14:52:37
山东畜牧兽医(2018年3期)2018-04-26 09:10:34
家庭科学·新健康(2016年9期)2016-10-25 16:05:12
中国卫生标准管理(2015年2期)2016-01-14 03:41:39
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:04
中国药业(2014年22期)2014-05-02 08:24:30
中国中医药现代远程教育(2014年14期)2014-03-01 04:27:20
