
Ni-Sn3O4/g-C3N4的制备及其光催化产氢性能研究
2022-12-09 04:31李洪吉王丹丹贺维韬王兴国
吉林师范大学学报(自然科学版) 2022年4期
王 昕,李洪吉,王丹丹*,贺维韬,王兴国
(1.吉林工程职业学院 粮油食品学院,吉林 四平 136000;2.吉林师范大学 工程学院,吉林 四平 136000;3.吉林师范大学 环境友好材料制备与应用教育部重点实验室,吉林 长春 130103;4.吉林师范大学 吉林省高校环境材料与污染控制重点实验室,吉林 四平 136000)
0 引言
在诸多半导体光催化剂中,锡基氧化物是一种低成本、易合成的材料,主要包括SnO、SnO2和Sn3O4.其中SnO和SnO2仅含有Sn2+或Sn4+,光催化性能较差.而Sn3O4是一种混合价氧化锡,相比于SnO和SnO2具有更高的光催化活性和更好的稳定性,受到了广泛的关注[1].虽然有很多关于Sn3O4的报道,但其光催化性能并不理想.X.Yu等[2]将Sn3O4与二维还原氧化石墨烯复合,改善了Sn3O4的光催化产氢性能,但产氢速率依然不是很高.由于单相Sn3O4很难有理想的光催化产氢活性,为了进一步提高Sn3O4的性能,R.Yang等[3]通过水热法制备了Ni掺杂的Sn3O4.Ni元素的引入加速了Sn3O4光生电子和空穴的分离,使Sn3O4的光催化性能得到提升.鉴于Ni-Sn3O4与g-C3N4的能带位置匹配[4].所以尝试将Ni-Sn3O4与g-C3N4复合并进行探究.
本文将Ni-Sn3O4与g-C3N4纳米片复合,构建2D/2D异质结构.通过XRD、FTIR、UV-Vis、SEM、TEM、氮气吸附等方法对材料进行表征,并研究复合材料的光催化产氢性能和反应机理.光催化产氢实验证明Ni-Sn3O4/g-C3N4复合材料的性能相比于g-C3N4得到提升,循环实验也证明了样品具有良好的稳定性.之后,通过PL和光电流测试对Ni-Sn3O4/g-C3N4复合材料光生电子和空穴的分离情况进行了探究.最后,根据测试结果提出了Ni-Sn3O4/g-C3N4复合材料中可能存在的电荷转移机制.
1 实验部分
1.1 试剂
尿素、氯化锡(Ⅱ)二水合物和氯化镍试剂购于麦克林试剂有限公司;柠檬酸三钠、氢氧化钠、三乙醇胺、无水乙醇、氯铂酸和硫酸钠购于国药集团化学试剂有限公司;硝酸购于辽宁泉瑞试剂有限公司.本实验中使用的所有化学试剂均为分析纯,在没有进一步纯化的情况下直接使用.在整个实验中使用的水为二次蒸馏水.
1.2 仪器
使用CuKα辐射D/max-2500铜阳极X射线粉末衍射仪(ARL EQUINOX 100,赛默飞世尔科技(中国)有限公司)在40 kV和200 mA条件下,记录样品的X射线衍射(XRD).使用红外光谱仪(Perkin-Elmer Spectrum One,美国铂金埃尔默公司)分析样品的红外光谱图.使用场发射扫描电镜(JEOL 7800F,日本JEOL公司)、透射电子显微镜(JSM-2100F,日本JEOL公司)观察记录样品的形貌并进行对比.通过全自动气体吸附仪(3H-2000PS1,美国康塔公司)分析样品比表面积(BET).利用紫外-近红外分光光度仪(Perkin-Elmer Lambda 900,日本岛津公司)记录粉末样品在300~800 nm范围的光学特性.在室温下,利用Renishaw inVia 激光拉曼光谱仪(325 nm,He-Cd激光器)记录样品的荧光光谱.
1.3 Ni-Sn3O4/g-C3N4材料的制备
1.3.1 g-C3N4纳米片的制备
将20 g尿素加入坩埚中,在马弗炉中以5 ℃/min的升温速率加热至550 ℃并保持4 h.冷却到室温后,将淡黄色固体分散至500 mL 0.1 mol/L的硝酸溶液中,在80 ℃下搅拌12 h.得到的固体粉末用去离子水和乙醇各洗涤3次,在80 ℃下干燥6 h.固体粉末再次放入马弗炉,以5 ℃/min的速率升温至500 ℃,保持2 h,得到g-C3N4纳米片,记为CN.
1.3.2 Ni-Sn3O4的制备
将0.169 g NiCl2·6H2O、1.128 g SnCl2·2H2O和3.676 g Na3C6H5O7·2H2O溶于25 mL去离子水中,分别搅拌、超声10 min,使其完全溶解分散.然后在不断搅拌下加入12.5 mL 0.2 mol/L NaOH溶液,超声使其均匀分散.将上述溶液转移到100 mL高压釜中,置于180 ℃的烘箱中加热12 h,待冷却至室温.所得粉末用去离子水和乙醇各洗涤3次,在80 ℃干燥10 h.
1.3.3 Ni-Sn3O4/CN的制备
将0.1 g g-C3N4纳米片分散于10 mL去离子水中,搅拌30 min.然后加入xmg(x=1,5,7,20)Ni-Sn3O4继续搅拌1 h.最后放入110 ℃的烘箱中加热4 h,得到的产物记为Ni-Sn3O4/CN-x(即Ni-Sn3O4/CN-1,Ni-Sn3O4/CN-5,Ni-Sn3O4/CN-7,Ni-Sn3O4/CN-20).
1.4 电化学性质研究
在3 mL无水乙醇中加入1 mg样品,超声30 min.取40 μL溶液滴涂在ITO导电玻璃的导电面,在常温黑暗条件下自然干燥24 h.电解液为0.5 mol/L的Na2SO4溶液.饱和甘汞电极、Pt电极、带有样品的ITO导电玻璃分别作参比电极、对电极和工作电极进行电化学测试.
1.5 光催化活性研究
将30 mg光催化剂加入到30 mL水溶液(10 vol%三乙醇胺)中,助催化剂为一定体积的H2PtCl6水溶液(3 wt% Pt).使用带有420 nm截止滤光片的300 W Xe灯(λ>420 nm)作为可见光光源.每隔30 min取一次样,通过气相色谱仪分析H2的产量.
2 结果与讨论
2.1 结构与形貌
图1为CN、Sn3O4、Ni-Sn3O4和Ni-Sn3O4/CN-x复合材料的XRD谱图.从图1中可以看到,石墨氮化碳三嗪单元的(100)晶面对应13.6°处的弱衍射峰,石墨氮化碳芳香族段层间堆积的(002)晶面对应27.4°处的强衍射峰[5].Sn3O4的衍射峰与XRD标准卡片(JCPDS card No.16-0737)一致,24.11°、27.05°、33.75°、37.27°、50.68°和51.72°处的衍射峰分别对应(101)、(111)、(-122)、(130)、(-311)和(-132)晶面[6].与Sn3O4相比,Ni-Sn3O4的衍射峰位置没有变化,但峰值强度明显高于Sn3O4.这可能是由于Ni的掺杂增加了Ni-Sn3O4的结晶度[7].与CN相比,Ni-Sn3O4/CN-x系列样品中相应CN的XRD衍射峰没有明显改变,初步表明Ni-Sn3O4的引入没有改变CN的化学结构.Ni-Sn3O4/CN-x系列样品中可以同时观察到属于CN和Ni-Sn3O4的衍射峰.随着Ni-Sn3O4/CN-x中Ni-Sn3O4含量的增加,在33.75°和51.72°处与Ni-Sn3O4相关的衍射峰强度逐渐增大,初步证明通过水热法成功将Ni-Sn3O4与CN复合.

图1 Ni-Sn3O4、Sn3O4、CN和Ni-Sn3O4/CN-x的XRD谱图
图2为CN和Ni-Sn3O4/CN-x样品的FTIR光谱.从图中可以看出,在810 cm-1处有一个明显的尖峰,属于g-C3N4中三嗪环的呼吸振动[8].在1 050~1 750 cm-1之间的几个吸收峰,归因于g-C3N4中CN杂环的伸缩振动[9].在g-C3N4中,未缩合的氨基和表面吸附的水分子引入了N—H和O—H键,形成了2 900~3 400 cm-1之间的宽峰[10].Ni-Sn3O4/CN-x的吸收峰与CN的吸收峰相似,进一步证明了Ni-Sn3O4的加入并没有改变CN的化学结构.
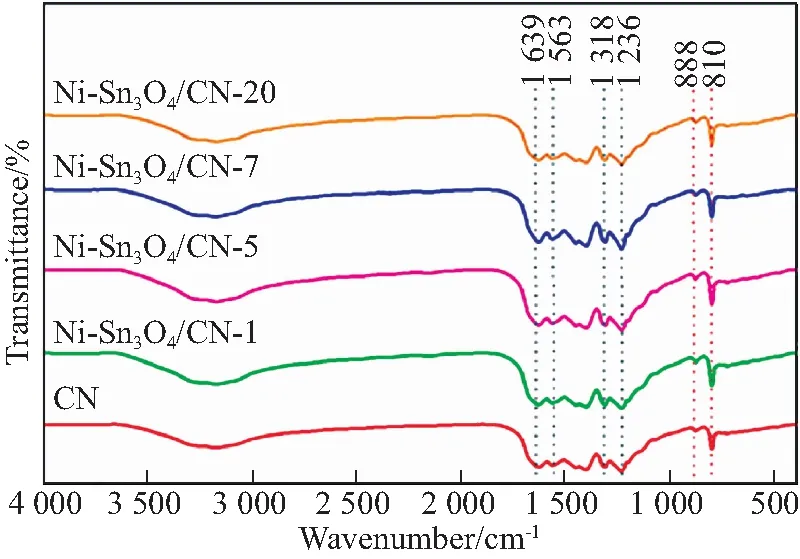
图2 Ni-Sn3O4、Sn3O4、CN和Ni-Sn3O4/CN-x的FTIR谱图
图3(A)、3(B)和3(C—D)分别是CN、Ni-Sn3O4和Ni-Sn3O4/CN-5的SEM图.如图3(A)所示,CN为二维的纳米片形貌.从图3(B)中可以看到Ni-Sn3O4为纳米花状形貌,花瓣部分是由许多小尺寸纳米薄片组合形成的.而且Ni-Sn3O4纳米花存在团聚现象.Ni-Sn3O4/CN-5的SEM如图3(C)和图3(D)所示,可以看到CN与Ni-Sn3O4紧密结合在一起,证明成功合成了Ni-Sn3O4/CN-5.图3(E—F)分别为Ni-Sn3O4/CN-5的TEM和HRTEM图像.从TEM图像中能够看到Ni-Sn3O4负载到CN上,二者结合紧密.HRTEM图像中显示出Ni-Sn3O4/CN-5具有间距为0.34 nm和0.36 nm的晶格条纹,与Ni-Sn3O4的(111)和(101)晶面一致,进一步证明成功合成了Ni-Sn3O4/CN-5复合材料[11].

图3 CN(A),Ni-Sn3O4(B)和Ni-Sn3O4/CN-5(C—D)的SEM图;Ni-Sn3O4/CN-5的TEM图(E)和HRTEM图(F)
2.2 比表面积和光学性质分析
为了了解样品的孔隙结构和比表面积,进行了BET(Brunauer-Emmett-Teller)测试.图4(A)为Ni-Sn3O4、CN和Ni-Sn3O4/CN-5的N2吸附-解吸等温线.三种样品的等温线均为Ⅳ型等温线,并带有H3型滞后环,说明所制备的样品具有介孔结构[12].图4(B)为Ni-Sn3O4、CN和Ni-Sn3O4/CN-5的孔径分布图.可以看出,CN、Ni-Sn3O4和Ni-Sn3O4/CN-5的主要孔径分别分布在30 nm、3 nm和30 nm.Ni-Sn3O4和CN复合之后,孔隙体积相比于CN有所增加,因此复合材料的比表面积会大于CN.表1为所制备材料的比表面积和孔结构数据.CN、Ni-Sn3O4、Ni-Sn3O4/CN-1、Ni-Sn3O4/CN-5、Ni-Sn3O4/CN-7和Ni-Sn3O4/CN-20的比表面积分别为40.70、21.70、43.50、44.77、43.18、42.78 m2/g.可以看出复合样品的比表面积大于CN的比表面积.因此,在光催化产氢反应过程中,Ni-Sn3O4/CN-x具有更多的活性位点、吸附更多的反应物[13].随着Ni-Sn3O4组分含量的增加,Ni-Sn3O4/CN-x样品的比表面积呈现出先增大后减小的火山型趋势.Ni-Sn3O4/CN-7和Ni-Sn3O4/CN-20比表面积的减小可能与Ni-Sn3O4在CN表面聚集过多有关[14].

图4 Ni-Sn3O4、CN和Ni-Sn3O4/CN-5的N2吸-解吸曲线(A)和孔径分布曲线(B)
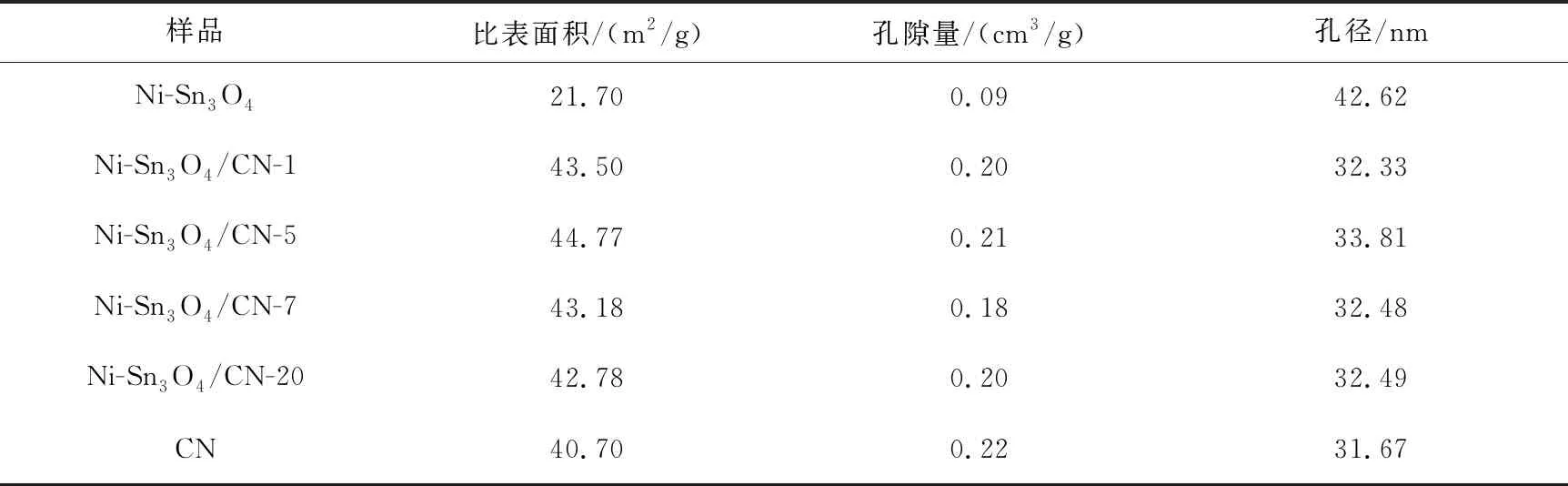
表1 Ni-Sn3O4、CN和Ni-Sn3O4/CN-x催化剂吸附数据总表
采用UV-Vis研究了Ni-Sn3O4、CN和Ni-Sn3O4/CN-x对光的响应能力.从图5(A)中可以看出,CN和Ni-Sn3O4的吸收边分别约为460 nm和520 nm.相比于CN,Ni-Sn3O4/CN-x样品对可见光吸收能力没有明显变化.利用Kubelka-Munk函数得到Tauc曲线,计算了Ni-Sn3O4和CN的带隙(Eg).如图5(B)所示,CN和Ni-Sn3O4的Eg分别为2.82 eV和2.67 eV.Ni-Sn3O4的带隙较小,这使得Ni-Sn3O4更容易被可见光激发[15].

图5 Ni-Sn3O4、CN和Ni-Sn3O4/CN-5的紫外可见漫反射图(A)和带隙图(B)
2.3 光催化活性分析
在可见光(λ>420 nm)照射下研究了Ni-Sn3O4、CN和Ni-Sn3O4/CN-x的光催化性能.从图6(A)中可以看到,各样品的产氢量随时间变化呈线性增长.其中,Ni-Sn3O4/CN-5的产氢量最高.图6(B)为各样品的光催化产氢速率图.Ni-Sn3O4和CN的产氢速率分别是12 μmol/(h·g)和1 393 μmol/(h·g).Ni-Sn3O4和CN复合后产氢速率得到明显提升,Ni-Sn3O4/CN-1、Ni-Sn3O4/CN-5、Ni-Sn3O4/CN-7和Ni-Sn3O4/CN-10的产氢速率分别为1 748、1 961、1 691、1 543 μmol/(h·g).其中Ni-Sn3O4/CN-5的产氢速率最高,分别是Ni-Sn3O4和g-C3N4的163倍和1.4倍.证明Ni-Sn3O4的引入对于提升CN的光催化产氢性能有很大的帮助.Ni-Sn3O4中Ni元素不仅能够调节能带结构和提高Sn3O4对可见光的吸收,还能够作为活性中心促进光生电子和空穴的分离和迁移速率,这也是Ni-Sn3O4/CN-5的光催化性能优于Ni-Sn3O4/CN-3的原因[16].相比于Ni-Sn3O4/CN-5,Ni-Sn3O4/CN-7和Ni-Sn3O4/CN-10的产氢速率明显降低,可能是由于过量的Ni-Sn3O4聚集使催化剂对光的吸收降低,影响了光生电子和空穴的生成.
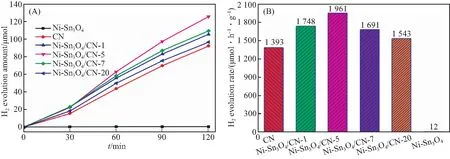
图6 Ni-Sn3O4、CN和Ni-Sn3O4/CN-x的光催化产氢量曲线(A)和光催化产氢速率图(B)
在与性能测试相同的条件下,进行了光催化循环实验.结果表明Ni-Sn3O4/CN-5光催化剂具有良好的稳定性.从图7(A)可以看出,在连续反应8 h后,Ni-Sn3O4/CN-5每2 h的总产氢量没有明显改变,说明Ni-Sn3O4/CN-5样品具有良好的稳定性.图7(B)为Ni-Sn3O4/CN-5在光催化反应前后的XRD谱图.Ni-Sn3O4/CN-5在光催化反应后的XRD衍射峰除强度相较于反应前略有降低外,位置没有明显偏移,进一步证明Ni-Sn3O4/CN-5的结构很稳定[17].
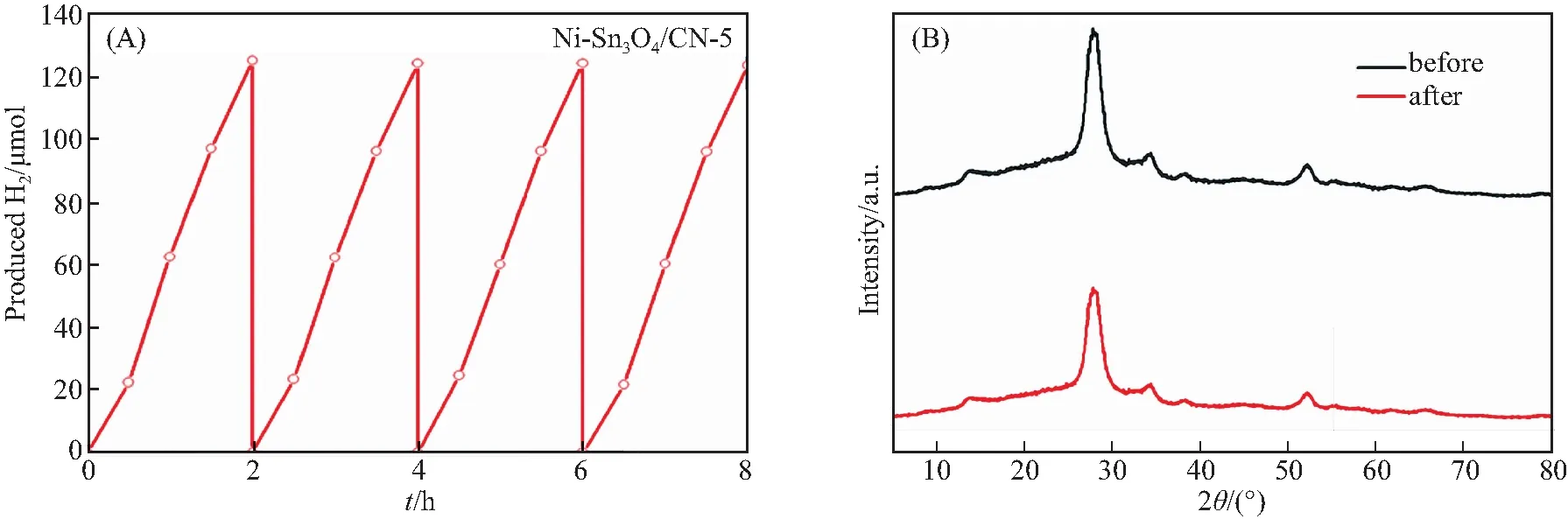
图7 4次循环后的光催化产氢量曲线(A)和光催化反应前后Ni-Sn3O4/CN-5的XRD对比图(B)
2.4 光电化学分析
在460 nm激发波长下测定了所有样品的PL光谱.图8为Ni-Sn3O4、CN和Ni-Sn3O4/CN-x的PL光谱.可以看到,CN在465 nm左右有较强的PL峰.Ni-Sn3O4/CN-x的峰值强度低于CN,说明引入Ni-Sn3O4可以加快光生电子和空穴的迁移和分离[18].在所有样品中,Ni-Sn3O4/CN-5的PL峰强度最弱,这意味着Ni-Sn3O4/CN-5的光生电子和空穴的分离效果更好,从而改善了Ni-Sn3O4/CN-5的光催化产氢性能[19].
通过电化学测试进一步研究了Ni-Sn3O4、CN和Ni-Sn3O4/CN-x中光生电子和空穴的迁移和分离.图9为样品的瞬态光电流曲线.在黑暗条件下,样品几乎没有响应.开灯后,样品的光电流密度迅速增加,关灯后,光电流密度迅速下降,说明所有样品对光非常敏感.在所有样品中,CN的光电流强度最低.相比于CN,复合材料的光电流强度均增强,说明Ni-Sn3O4的加入有助于光生电子和空穴的分离[20-21].在所有复合材料中,Ni-Sn3O4/CN-5的光电流强度最强.
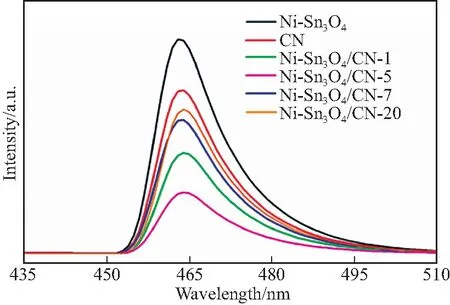
图8 Ni-Sn3O4、CN和Ni-Sn3O4/CN-x的PL谱图
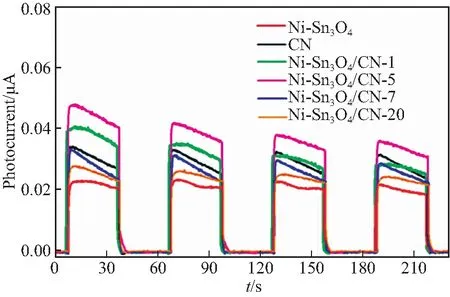
图9 Ni-Sn3O4、CN和Ni-Sn3O4/CN-x的光电流
2.3 光催化机理分析
根据Ni-Sn3O4和CN的能带结构排布,Ni-Sn3O4和CN之间所形成的异质结构可能为Ⅱ型或S型异质结.假设Ni-Sn3O4和CN之间形成的是Ⅱ型异质结,电子将从CN向Ni-Sn3O4转移,而Ni-Sn3O4的导带电势比CN更正,对氢质子的还原能力比CN弱,这与Ni-Sn3O4/CN-5光催化产氢性能的提升不符.因此,Ni-Sn3O4和CN之间形成的应是S型异质结.Ni-Sn3O4/CN-5的光催化机理如图10所示.由于CN的导带和价带电势高于Ni-Sn3O4,会有电子从CN转移到Ni-Sn3O4[22].在Ni-Sn3O4和CN的界面处,CN界面失去电子,带正电荷,Ni-Sn3O4界面得到电子,带负电荷.电子的转移会导致在两种材料的界面处形成一个内建电场.同时,CN的带边因失去电子而向上弯曲,而Ni-Sn3O4的带边因得到电子而向下弯曲[23].在可见光照射下,CN和Ni-Sn3O4价带上的电子被激发到导带.在内建电场和带边弯曲的共同作用下,在加速了一些电子(Ni-Sn3O4的CB)和空穴(CN的VB)的复合的同时,阻止了一些电子(CN的CB)和空穴(Ni-Sn3O4的VB)的复合.S型的电子转移机制加速了Ni-Sn3O4上电子和CN上空穴的复合,使CN上的电子和Ni-Sn3O4上的空穴被很好的保留下来并参加接下来的氧化还原反应,这使Ni-Sn3O4/CN-5的光生电子和空穴得到很好的分离.此外,内建电场的形成改善了催化剂的氧化还原能力,进一步提高了Ni-Sn3O4/CN-5的光催化产氢性能.其次,Ni-Sn3O4/CN-5中Ni元素作为电荷转移的活性位点,进一步促进了光生载流子的迁移,这也是Ni-Sn3O4/CN-5光催化性能优于Ni-Sn3O4/CN-3的主要原因.
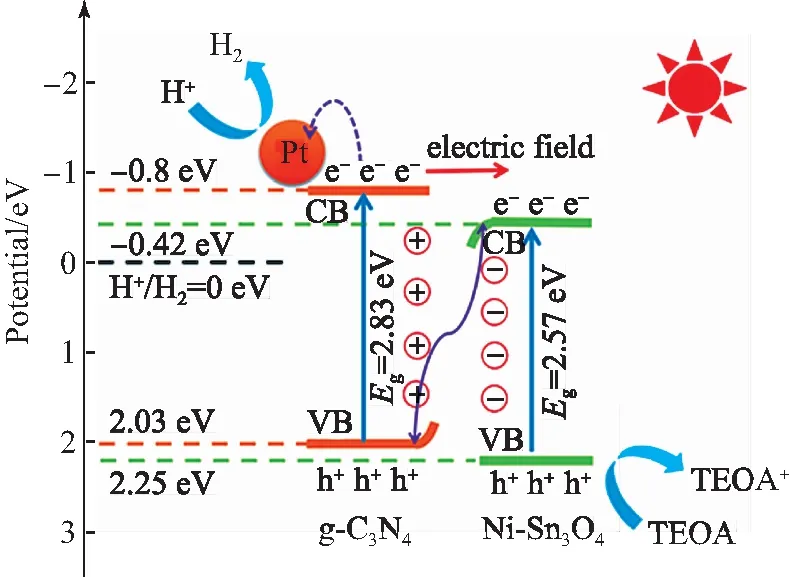
图10 Ni-Sn3O4/CN-5的光催化产氢机理
3 结论
本文通过水热法将Ni-Sn3O4和g-C3N4复合制备了一系列不同比例的样品(Ni-Sn3O4/CN-1、Ni-Sn3O4/CN-5、Ni-Sn3O4/CN-7、Ni-Sn3O4/CN-10).通过XRD、FTIR、SEM和TEM测试证明了材料的成功合成.在可见光照射下,对所合成材料的光催化产氢性能进行了测试.结果表明,所有复合材料的光催化产氢速率均高于g-C3N4.其中,Ni-Sn3O4/CN-5的光催化性能最好,产氢速率达到1 961 μmol/h·g.四次循环实验和催化剂在光催化产氢反应前后的XRD测试证明了复合材料的稳定性良好.光电测试结果证明,Ni-Sn3O4/g-C3N4具有更好的光生电子和空穴的分离效率,是复合材料光催化产氢性能提升的主要原因.
猜你喜欢
科学之友(2022年11期)2022-11-03
工业水处理(2022年6期)2022-06-23
力学学报(2022年5期)2022-06-16
车用发动机(2021年5期)2021-10-31
石油化工高等学校学报(2021年3期)2021-07-15
河南科学(2020年7期)2020-09-10
化工设计(2020年2期)2020-05-01
物理化学学报(2019年8期)2019-09-03
陕西科技大学学报(2018年1期)2018-01-11
郑州大学学报(理学版)(2017年1期)2017-04-07
