
Toward accurate and efficient dynamic computational strategy for heterogeneous catalysis: Temperature-dependent thermodynamics and kinetics for the chemisorbed on-surface CO
2022-12-07 08:27:04JunChenTanJinYihuangJiangTonghaoShenMingjunYangZheNingChenb
Chinese Chemical Letters 2022年11期
Jun Chen, Tan Jin, Yihuang Jiang, Tonghao Shen, Mingjun Yang,Zhe-Ning Chenb,,d,∗∗
a Fujian Science & Technology Innovation Laboratory for Optoelectronic Information of China, Fuzhou 350108, China
b State Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, China
c State Key Laboratory of Physical Chemistry of Solid Surfaces, Fujian Provincial Key Laboratory of Theoretical and Computational Chemistry, Xiamen University, Xiamen 361005, China
d University of Chinese Academy of Sciences, Beijing 100049, China
e MOE Key Laboratory of Computational Physical Sciences, Department of Chemistry, Fudan University, Shanghai 200433, China
f Shenzhen Jingtai Technology Co., Ltd.(XtalPi), Fubao Community, Shenzhen 518045, China
Keywords:Dynamic strategy Temperature-dependent thermodynamics Statistical sampling Neural networks potential energy surface Operando simulation
ABSTRACT Computational tools on top of first principle calculations have played an indispensable role in revealing the molecular details, thermodynamics, and kinetics in catalytic reactions.Here we proposed a highly efficient dynamic strategy for the calculation of thermodynamic and kinetic properties in heterogeneous catalysis on the basis of efficient potential energy surface (PES) and MD simulations.Taking CO adsorbate on Ru(0001) surface as the illustrative model system, we demonstrated the PES-based MD can efficiently generate reliable two-dimensional potential-of-mean-force (PMF) surfaces in a wide range of temperatures, and thus temperature-dependent thermodynamic properties can be obtained in a comprehensive investigation on the whole PMF surface.Moreover, MD offers an effective way to describe the surface kinetics such as adsorbate on-surface movement, which goes beyond the most popular static approach based on free energy barrier and transition state theory (TST).We further revealed that the dynamic strategy significantly improves the predictions of both thermodynamic and kinetic properties as compared to the popular ideal statistic mechanics approaches such as harmonic analysis and TST.It is expected that this accurate yet efficient dynamic strategy can be powerful in understanding mechanisms and reactivity of a catalytic surface system, and further guides the rational design of heterogeneous catalysts.
The continuous demand for the sustainable development of our society requires the development of more active, more selective, and less expensive catalytic processes to eventually solve energy and environmental problems.Revealing the catalytic molecular mechanism and their corresponding statistical thermodynamic information is not only a central issue in catalytic science, but also provide a useful guide for the further rational design of the effi-cient catalytic processes [1–4].Although an enormous amount of experimental effort has been conducted for molecular mechanism in heterogeneous catalysis [5–8], there is still experimental technical difficulties of obtaining molecular information directly.Theoretical and computational tools thus have become an indispensable approach for revealing the molecular details as well as the thermodynamics in catalysis.
Nowadays, theoretical calculations become really common in the field of catalysis.This, aligned with the rapid development in theoretical methods as well as the enormous growth in computational power.The static computational strategy is the most common approach in theoretical catalysis [2], in which only the limited stationary states at zero temperature are localized.The thermodynamics at specific conditions is computed by using the partition functions of ideal models.The ideal gas expressions are typically used for translational and rotational contributions of free molecules while the harmonic approximation is used for vibrational partition functions.This approach is computationally economical and thus has been widely used in theoretical catalysis.However, the reliability of this static approach depends on whether the realistic conditions significantly deviates from the ideal models [2].
The value of computational results for catalysis lies on whether the reliable computational thermodynamics at the specific condition could be obtained in theoretical approach.Notably, reaction temperature often plays a significant role in the activity as well as the selectivity for a catalytic reaction [9–13].Accurate calculation of the temperature-dependent thermodynamics in catalysis is thus critical for revealing the correct molecular mechanism and further guiding the rational design of more efficient catalytic processes.However, the degree of deviation between the realistic conditions and ideal models should closely relate to the temperature, which makes the reliability of temperature-dependent thermodynamics from the common static computational approach be doubted.Therefore, development an alternative computational strategy for theoretical catalysis that going beyond the common static computational strategy becomes necessary for calculating the reliable temperature-dependent thermodynamics.
Molecular dynamics (MD) is a well-tested approach to obtain the thermodynamics of the target system by going beyond static stationary states and sampling phase space more broadly[2,14,15].However, the computational cost for this dynamic computational strategy is significantly higher than the common static computational strategy.Catalytic processes generally involve breaking and formation of chemical bonds, clearly implies the necessity of employingab initioelectronic structure methods, instead of the more economical classical force fields, in the simulations,to provide a reasonably accurate description of the complicated electronic/chemical interactions.Ab initioMD (AIMD) simulation is thus an appropriate choice for studies of catalysis, however,AIMD simulation achieves a reasonable length to provide meaningful thermodynamics is still challenging.Therefore, the most important aspect for using dynamic computational strategy in catalysis is to overcome the limitation of time scales in AIMD simulations.On one hand, proper enhanced sampling approaches could be introduced to allow an accelerated search in the phase space and thus fast thermodynamic properties evaluation.On the other hand, development of an efficient and accurate strategy to replace the directab initiocalculations on system’s energy and force is another approach to achieve a converged sampling more efficiently.
Various enhanced sampling strategies have been developed so far for accelerating phase space sampling, including but not limited to the approaches of umbrella sampling (US) [16], replica exchange[17,18], simulated tempering [19,20], transition path sampling [21],metadynamics (MTD) [22,23], adaptive biasing force [24], temperature accelerated molecular dynamics (TAMD) [25–28], and integrated tempering sampling (ITS) [29,30], as well as some strategies of combining two enhanced sampling methods, such as ITSMTD method developed by Yanget al.[31], ITS-US and ITS-TAMD methods developed by us [32,33].Recently, the enhanced sampling methods were combined with AIMD simulations to solve some issues in heterogeneous catalysis [2,34,35].It’s worth mentioning that we have provided a dynamic computational strategy by combining ITS method and AIMD simulation for the temperaturedependent thermodynamics of CO diffusion on Ru(0001) surface[35].Although the enhanced sampling method indeed significantly improves the sampling efficiency, the computational cost is still awfully expensive for achieving a converged sampling.Limited thermodynamic information only at three temperature was gave in our previous ITS enhanced AIMD simulations.Therefore, we have been aware of that such enhanced AIMD based dynamic computational strategy is still too expensive to face the realistic issues in catalysis.Further reducing the cost in calculation of system’s energy and force is necessary.
Construction of the accurate global potential energy surface(PES) could avoid the expensive directab initiocalculations.Actually, an accurate PES is essential for the theoretical research in chemical reaction dynamics.In the past few decades, development of fitting techniques ofab initioelectronic structure data has made the construction of accurate global PESs possible for multidimensional molecular systems [36].More recently, the artificial neural network (NN) methods have displayed powerful in construction of the complex PES, which promotes the wide utilization of NN PES on the chemical reaction dynamics for multidimensional molecular systems [36–39].The NN PES, especially the development of atomistic neural network (AtNN) architecture [40], also gives hope for dealing with the more realistic chemical issues [41–46].Actually,some limited MD simulations based on NN PES have appeared in the material and catalysis science [47].
The adsorption and activation of CO molecule on a transition metal surface is often a critical elementary step for the catalytic conversion of CO into the high added-value chemicals, such as Fischer-Tropsch reaction [48–50], CO2reduction [51–53], and coal to ethylene glycol [54–56].Given the fact that there exist many active sites on the surface, presumably with different binding affinity to the substrates, intermediates, as well as transition states,a proper description of the adsorbate adsorption and movement between different sites on surface is thus clearly important for a correct understanding of the catalytic process.It is worth noting that CO molecule adsorbed on a transition metal surface is a prototype system for molecular chemisorption [57], and the diffusion of CO molecule on metal surfaces in fact involves breaking and formation of metal-carbon bonds, very much like a typical chemical reaction process.This fact clearly implies the economical classical force fields cannot make a reliable description for the adsorption and movement for CO on surface.
In this contribution, we proposed an accurate and efficient dynamic strategy for the temperature-dependent thermodynamics of heterogeneous catalysis, using CO adsorbate on Ru(0001) surface as the illustrative model catalytic system.It should be noted that a six-dimensional PES of this system has been reported by Füchselet al.[58].However, it is predicted on their PES that the adsorption mode onhcpsite is unstable and the CO will move to thetopsite without a barrier, which is contrary to the widely accepted viewpoint that bothtopandhcpare stable adsorption modes [59,35].Here a new 6D PES based on NN was constructed beforehand instead of the directab initiocalculations during MD trajectory integrations.For comparison, a high-dimensional PES based on Embedded Atom NN (EANN) [44] was constructed and MD simulations were conducted with surface atoms relaxed.The proposed dynamic strategy obtained the accurate temperature-dependent thermodynamics efficiently and generated the reliable smooth twodimensional potential-of-mean-force (2-D PMF) surfaces in a wide range of 300–900 K.By proper considering the thermodynamic contribution from the adsorbate in-plane motions that was missed when generating the 2-D PMF surfaces, the free energy difference as well as the diffusion barrier between the two stable adsorption sites (topandhcp) were unexpected found to increase rather than reduce with temperature raising.Benefit from the high efficiency for our proposed dynamic strategy, the temperature-dependent kinetics for the adsorbate on-surface movement can be calculated directly from the converged sampling trajectories, which provides a chance to evaluate the validity of transition state theory (TST) that is the most common approach for kinetics calculation in theoretical catalysis.Our calculations indicated there is significant quantitatively deviation for TST in the adsorbate on-surface kinetics.Finally, we made a careful comparison between the proposed dynamic computational strategy and the common static computational strategy, and further revealed the limitation of static computational strategy in temperature-dependent thermodynamics.Our study clearly demonstrates the efficiency and reliability of the dynamic computational strategy based on efficient PES and MD simulations, which is expected as a powerful tool for the statistical thermodynamics and kinetics calculation in catalysis, in conjunction with the proper enhanced sampling approaches when necessary.
For the illustrative model catalytic system of chemisorbed onsurface CO, theab initiocalculations were based on Perdew-Burke-Ernzerhof (PBE) functional [60] with the VASP (Viennaab initioSimulation Package) code [61,62], using the projector-augmentedwave (PAW) [63] method together with plane-wave basis sets to describe the electron-ion interactions.Integration over the Brillouin zone was performed by using the Monkhorst-Pack scheme[64] with 3×3×1k-points.The 6D PES, in which the CO molecule was scattered on the rigid Ru(0001) surface, was constructed with NN fitting to a data set sampled using an efficient configuration selection scheme [65].For comparison, a high-dimensional PES, with 3 layers of surface atoms relaxed, was constructed using the EANN approach [44] developed by Jiang and co-workers.The EANN potential is an improved variation of Atomic-NN potential [40], using the embedded-atom model [66] to describe the local environment of atoms.Molecular dynamics (MD) were simulated in the canonical ensemble with the Nosé-Hoover thermostat [67,68] for temperature controlling, and the velocity-verlet algorithm [69,70]to integrate the equation of motion, using energies and forces directly from the NN PES.2-D PMF surfaces were generated from the MD trajectories.It is worth noting that the thermodynamic contribution of the two in-plane modes of adsorbate is missed in a 2-D PMF surface scheme.Therefore, relative free energy for the adsorbate on a specified on-surface site was evaluated not only on a local minimum point but also within a region near degeneracy in free energy.Similarly, the free energy barrier for adsorbate diffusion was evaluated by considering the occupation probability of adsorbate on a 2-D area of the PMF with the close free energy.The rate constant of barrier crossingkis evaluated by TST from the free energy barrier.In addition, the rate constants have also been directly calculated from the diffusion time of MD trajectories.As a comparison, the common static computational strategy was also conducted for thermodynamic properties evaluation.More detailed computational methods can be found in supplementary material.
The highly efficient in the current NN PES for calculating the interaction between the adsorbate and surface makes it possible to achieve the converged sampling by a long length simulation.The temperature-dependent thermodynamics were obtained from MD simulations for a total time of 3.5 μs at each of seven different temperatures, ranging from 300 K to 900 K.The obtained 2-D PMF surfaces for CO adsorbate on rigid Ru(0001) are illustrated in Fig.1 and Fig.S5.The PMF surfaces clearly show the existence of two distinct thermodynamically stable CO binding sites,topandhcp.Thetopsite is more favored thanhcpsite at all the simulated temperatures.This agrees with the previous experimental and theoretical results [71,72] and confirms the reliability of this newly reported PES.As shown in Fig.1 and Fig.S5, with increasing temperature, the 2-D PMF surfaces grow flatter and flatter, which means the distribution of CO adsorbate at different surface sites becomes more and more uniform, agreeing with the observed increased presence of CO at higher coordination sites with temperature raising in the previous spectroscopic experiments [73–75].
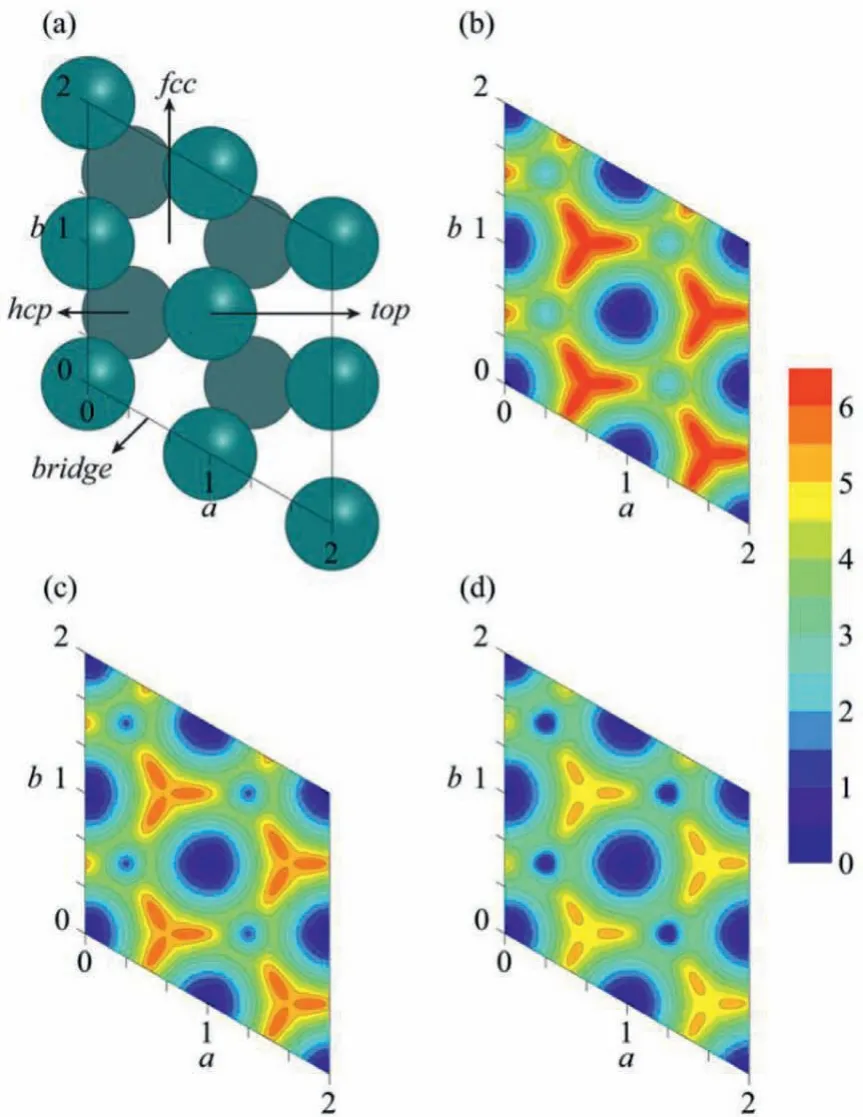
Fig.1 .Model of Ru(0001) surface and 2-D PMF surfaces of CO adsorbate on the Ru(0001) surface along the fractional coordinates a and b of carbon atom on the surface plane.(a) Ru(0001) surface with a 2×2 supercell in the lateral directions;(b) PMF at 300 K; (c) PMF at 600 K; (d) PMF at 900 K.
The PMF surfaces show that there are two stable CO binding sites,topandhcp, on the Ru(0001) surface.First, we quantitatively describe the binding free energy difference between the two sites by only considering the local minimum point ontopandhcpsites,respectively.As shown in the blue line of Fig.2a (noted as the“PMF point” approach), the calculated binding free energy difference betweentopandhcpsites becomes smaller as temperature increases, in line with the observed PMF surfaces.However, the thermodynamic contribution of in-plane modes of adsorbate has been missed if only the local minimum point in 2-D PMF surface was considered.Instead, the missed contribution from the in-plane modes could be largely included when the occupation probability was calculated by considering CO adsorbate in a region other than a single local minimum point.Therefore, the relative adsorption free energy on a stable binding site was evaluated based on the local minimum point with additional near degeneracy region(within 1 kcal/mol) around the point.As shown in Fig.1, the near degeneracy region intopsite is obviously larger than that inhcpsite, suggesting the more freeness for CO adsorbate movement on thetopsite.The distribution of CO adsorbate attopandhcpregions still becomes more and more uniform with the rising of temperature (Table S1 in Supporting information).However, it is unexpected to find that their variation of relative free energy exhibits an opposite trend.As shown in the red line of Fig.2a (noted as the “PMF region” approach), the free energy difference betweentopandhcpregions increases as temperature raising, in contrary to the intuition.Further analysis of the contribution from enthalpy and entropy revealed that, as shown in Fig.2b, the entropic effect plays an important role in the temperature-dependent free energy change.As the more freeness for adsorbate movement on thetopsite, adsorption of CO molecule ontopsite should be facilitated by entropic effect, resulting in the observed unexpected temperaturedependent thermodynamic properties.
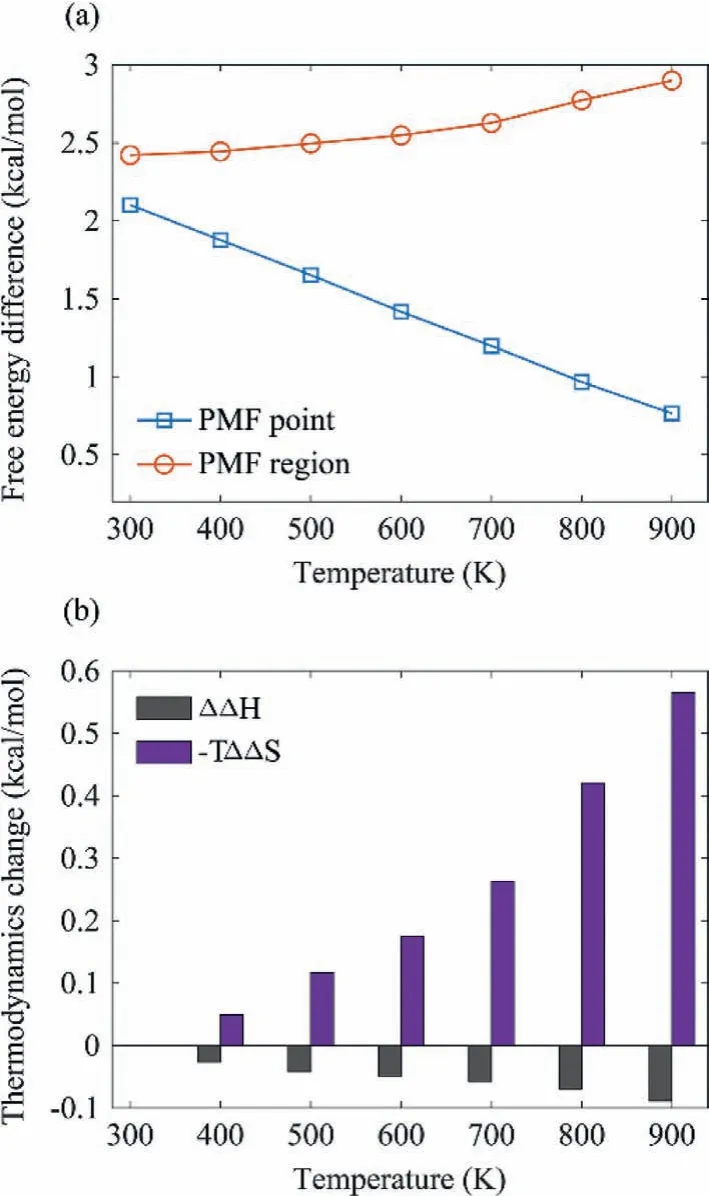
Fig.2 .Temperature-dependent (a) free energy difference and (b) corresponding contribution from enthalpy and entropy between top and hcp sites calculated based on PMF surfaces.
Diffusion of adsorbates is the primary means for accomplishing two crucial events required for catalysis: the encounter of different reaction partners to form the reactant complex and the arrival at an active site which provides strong affinity for the active intermediate of the reaction.The PMF surfaces clearly show significant barriers separating different binding sites.The diffusion of CO adsorbate should quite likely follow a typical jumping-among-minima behavior, making an elementary process in surface catalysis.The diffusion kinetics could be obtained based on the calculated free energy barrier in conjunction with TST, likely the most common approach in theoretical catalysis.Moreover, we would like to mention another approach to calculate the diffusion kinetics based on the dynamic protocol proposed here, that is, calculating the diffusion rate constant (k) directly from the average diffusion time of MD trajectories jumping from one adsorption site to another.Based on adequate sampling, such approach undoubtedly possesses a more accuracy since the intrinsic error of TST can be properly avoided.As mentioned above, definition of a proper region around the local minimum point is necessary for evaluating the thermodynamics for a specified binding site.Similarly, definition of a proper region around the saddle point is necessary for calculating the kinetics.Accordingly, the diffusion barrier can be calculated based on the occupation probability difference of CO adsorbate between the defined TS region and the corresponding local minimum region.The calculated diffusion barriers based on PMF surfaces for CO adsorbate moving fromtoptohcpregion were illustrated as gray bars in Fig.3a.The corresponding diffusion rate constants (k)can be further evaluated based on TST and were illustrated as a gray line in Fig.3b, noted as the “PMF region+TST” approach.As a comparison, the free energy differences between TS andtopsites displayed on 2-D PMFs were shown as black bars in Fig.3a.On the other hand, the diffusion rate constants were calculated directly from the average diffusion time of MD trajectories, and illustrated as a blue line in Fig.3b, noted as the “MD” approach.The corresponding diffusion free energy barriers can be backstepped based on TST and illustrated as blue bars in Fig.3a, noted as the“MD+TST” approach.Our results showed that the diffusion barriers calculated by the “PMF region” and “MD+TST” approaches both increase with the rise of temperature, in consistent with the variation of relative free energy betweentopandhcpregions with temperature change shown in Fig.2a.However, there is significant difference in high temperature limit for the calculated kinetics by these two approaches, which can be mostly attributed to the limitation of TST.As shown in Fig.3a and Table S2 (Supporting information), we took the absolute percentage difference (APD) to evaluate the deviation in the backstepped free energy barrier from TST based on the calculatedkby the “MD” approach.As shown,the APD is as high to 30.4% atT=900 K, and is showed to reduce along with the decreasing of temperature.When temperature drops below 500 K, the APD is reduced to within 10%.Meanwhile,As shown in Fig.3b and Table S2, the deviation ofkbased on the free energy barrier and TST was also evaluated.As shown, the TST underestimated thekby 38.8% at 300 K, and increased dramatically to 184.4% when temperature is raised to 900 K.In contrast, the free energy differences (noted as “PMF point” approach in Fig.2a) show an opposite trend with the change of temperature, mainly due to the lack of two in-plane modes considered in free energy calculations.It is also found in Fig.3b that, different to TST rates based on PMF barriers, the directly calculated rates from diffusion time of MD trajectories show an obvious non-Arrhenius property, which should be close to the practical motif of an elementary process in heterogeneous catalysis [76,77].
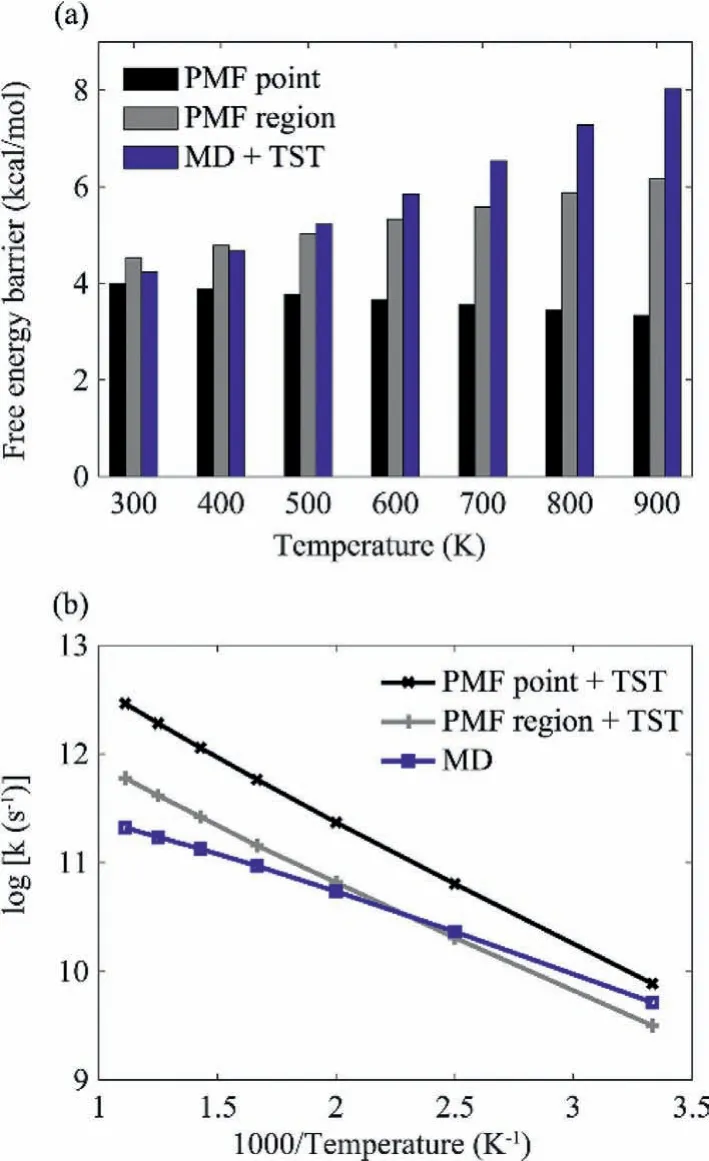
Fig.3 .Temperature-dependent (a) free energy barriers and (b) rate constants for CO adsorbate diffusion from top to hcp region calculated based on PMF surfaces and diffusion time of MD trajectories, respectively.
The static computational strategy, which is based on the relative potential energy and harmonic vibrational frequencies of an optimized transition state, is the most common strategy in theoretical catalysis.However, its reliability depends on the degree of the realistic condition’s deviation from the ideal models.Here the benchmark results from dynamic computational strategy provide us a chance to go beyond the ideal model so that reach a higher accuracy compared to statistic thermodynamic properties.As shown in Fig.4 and Table S3 (Supporting information), although the variation tendency for the temperature-dependent thermodynamics between static and dynamic computational strategies is consistent,the static strategy significantly underestimates both the relative free energy and diffusion barrier betweentopandhcpregions.Our calculations indicated the APD for the calculated relative free energy from static strategy against that from dynamic strategy are in the range of 30% to 37%.The difference of calculated free energy barriers shown in Fig.4a will introduce APD of 159.4% to 265.6%in the diffusion rate, as listed in Table S3.Moreover, the deviation for static strategy is shown to increase along with the raising of temperature.At the high temperature limit of 900 K, result from static strategy cannot even reach chemical accuracy.These findings suggest that, there is indeed significant deviation for description of the adsorption and movement for adsorbate on surface by using the common static strategy, especially in a high temperature.
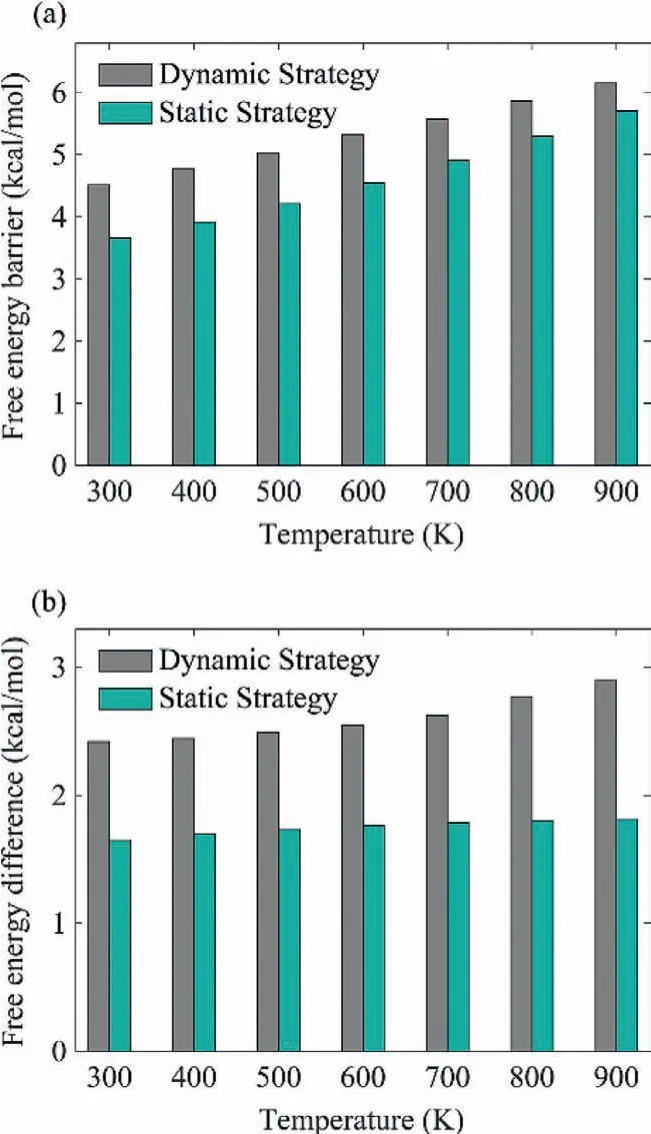
Fig.4 .Temperature-dependent (a) free energy barrier and (b) free energy difference for CO adsorbate diffusion from top to hcp region calculated based on dynamic strategy (the “PMF region” approach) and static strategy, respectively.
Now we have to indicate that, in the computational investigation on catalysis, the static computational strategy is the most common approach, in which the limited stationary states at zero temperature are localized and subsequently based on the partition functions of ideal models to obtain the thermodynamics at the realistic experimental conditions.Therefore, the reliability of static computational strategy depends on whether the ideal models are deviated from the realistic conditions.In comparison, MD simulations is a well-tested approach to obtain the thermodynamics as well as the kinetics of the target system by going beyond ideal models.The reliability of the dynamic computational strategy in statistical thermodynamics depends on whether the converged sampling can be achieved.
The dynamic computational strategy based on AIMD simulations has been used to face some issues in heterogeneous catalysis[2].However, the expensive computational cost in AIMD simulations greatly limited its wide applications.The efficiency of analytical PES provides an accurate and efficient computational approach by avoiding most expensiveab initiocalculations.Therefore, a dynamic computational strategy by combination of efficient PES and MD simulations has been proposed here for efficient evaluation of temperature-dependent thermodynamics for heterogeneous catalysis.We have constructed a six-dimensional PES for CO adsorbate on Ru(0001) surface, which can fully reproduce the potential energy and gradients atab initiolevel.Base on it, converged sampling by MD simulation has been carried out for the temperaturedependent thermodynamics of CO adsorption and diffusion on the surface.Benefit from our highly efficient dynamic computational strategy, the temperature-dependent thermodynamics have been obtained from a total of 3.5 μs long MD simulations at each of seven temperatures, that is 300–900 K.
We obtained the smooth 2-D PMF surfaces for CO adsorbate on Ru(0001) surface at 300–900 K (Fig.1 and Fig.S5), displaying the intuitive pictures of temperature-dependent thermodynamics.However, the thermodynamic contribution of adsorbate in-plane motion is missed when the phase space is projected to a 2-D PMF surface.We thus proposed here using a properly defined region other than a single local minimum (or saddle) point to include the contribution of adsorbate in-plane motion.Our results unexpectedly showed that the relative free energy betweentopandhcpsites increases rather than reduces as the temperature raising, being contrary to the intuition (Fig.2).The PMF surfaces in Fig.1 and Fig.S5 obviously suggest that the free energy curve intopsite is flatter than that inhcpsite, in agreement with our previous results that the frequency of the two in-plane modes is lower in thetopsite [35] and further suggests thetopsite has less steric interactions.Therefore, the in-plane motion is easier in thetopsite, which makes the adsorption and movement of CO molecule on top site is more facilitated by entropic effect, resulting in the observed counterintuitive temperature-dependent thermodynamic properties.
In the common computational treatment in theoretical catalysis, the reaction kinetics are mostly obtained from the combination of calculated free energy barrier and TST.Our proposed efficient dynamic computational strategy can not only provide the free energy barrier by a proper statistical protocol, but also calculate the diffusion rate constantkdirectly from the average diffusion time of MD trajectories.It means the evaluation of kinetics is not necessary to rely on the TST in our newly provided dynamic computational strategy, which provides a benchmark to evaluate the validity of TST.As shown in Fig.3 and Table S2, there is significant deviation for the calculated kinetics in TST, indicating the limitation of TST for reaction kinetics, or at least for the on-surface diffusion processes.Our calculations showed the deviation for TST is particularly significant at high temperature.Such as, the APD for diffusion barrier is as high to 30.4%, and the rate constantkis even overestimated by an APD value of 184% at 900 K.The deviation of TST is shown to reduce along with the drop of temperature.It has been proved again that TST is improper for the kinetics of low barrier process, further revealing the necessity for using dynamic computational strategy in direct evaluation of the more accurate catalytic kinetics in quantitative.It is also suggested that the non-Arrhenius behavior of an elementary process, which should be universal in dynamics, can only be accessed by the direct computation ofkfrom the average diffusion time of MD trajectories.
Our proposed dynamic computational strategy also provides a chance to evaluate the reliability of the common static computational strategy.As shown in Fig.4 and Table S3, there is significant deviation for static computational strategy in quantitative, in which the APD for the calculated thermodynamics and kinetics are within the range of 30% to 37% and 157% to 266%, respectively, revealing the limitation of ideal statistical models.We are pleased to find that there is consistent variation tendency for the temperaturedependent thermodynamics between static and dynamic computational strategies, indicating the applicability of static computational strategy in qualitative for such simple case.However, the deviation of static computational strategy is expected to be further increased when faced with the more complex realistic catalytic systems.The dynamic computational strategy thus become necessary if the quantitatively accurate description for catalytic processes and the further catalyst rational design are required.
Here we have to indicate that the rigid surface model was used at current stage.The simulated results would deviate from the realistic conditions to some extent, especially at the relatively high temperature.However, we focus on the performance of dynamic computational strategy in description of the adsorbate molecule currently, a rigid surface is thus a simple but clear representative theoretical model.Based on the newly constructed highdimensional PES, we got some preliminary results when two layers of surface Ru atoms were relaxed in MD simulations, and displayed in Fig.S7 and Table S4 (Supporting information).The PMF surfaces based on rigid surface model and flexible surface model seem similar in appearance, while the quantitative difference between them is obvious.Thefccsite, which is an unstable adsorption site at a rigid surface, becomes a local minimum when surface Ru atoms are relaxed.Moreover, the temperature-dependent diffusion barriers are quite different from the results of the rigid surface model,probably due to the energy transfer between the adsorbate and the surface phonons.One more issue is found when the degrees of freedom for surface atoms motion are considered.The definition of the different sites on surface, such astopandhollowsites,is no longer as clear as in the current rigid surface model since the effect of surface atoms motion.Investigation by using the flexible surface model for further approaching the realisticoperandoconditions based on the more general atomistic NN architecture is ongoing.
In summary, we have proposed an efficient dynamic computational strategy for catalysis based on the combination of effi-cient PES and MD simulations.Based on a six-dimensional analytical PES, longtime MD simulations have been carried out for the temperature-dependent thermodynamics of adsorbate adsorption and movement on surface.The smooth and reliable 2-D PMF surfaces at seven different temperatures (from 300 K to 900 K) have been obtained, and abundant temperature-dependent thermodynamic properties have archived.To the best of our knowledge, this is the first report of temperature-dependent thermodynamics and kinetics for a chemisorbed molecule on transition metal surface from MD simulations on a PES.
The temperature-dependent thermodynamics as well as the diffusion kinetics for adsorbate on-surface adsorption and movement have been explored by this dynamic computational strategy.We are unexpected to find that the calculated free energy difference and diffusion barrier between the two stable binding sites (topandhcp) increase other than reduce along with the temperature raising, contrary to the common intuition.In addition, our dynamic computational strategy provides an efficient approach to evaluate the reaction kinetics directly, and also provides a benchmark to evaluate the applicable of TST that is the most common computational tool for obtaining reaction kinetics in theoretical catalysis.Our results also showed that there is significant deviation for TST in on-surface diffusion kinetics.
Our dynamic computational strategy also provides a chance to evaluate the reliability of common static computational strategy.Our results indicated there is significant deviation of static computational strategy, although the static computational strategy performs a right variation tendency with temperature change in qualitative for this selected simple case.It can be expected that the deviation of static computational strategy would be further increased when faced with the more complex realistic catalytic systems.These findings demonstrated the necessity for usage of dynamic computational strategy to obtain the quantitatively accurate information in catalysis.Our study clearly indicated that the dynamic computational strategy based on the combination of efficient PES and MD simulations is readily available as an efficient and reliable approach to study the thermodynamics and kinetics of catalysis.By further combination of the advanced density functional approximations [78–81] as well as the proper enhanced sampling protocols[31–33], the proposed dynamic computational strategy is expected to open an avenue for accurate and efficientoperandocomputational modeling for heterogeneous catalysis.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work is financially supported by Fujian Science & Technology Innovation Laboratory for Optoelectronic Information of China(No.2021ZR109), the National Natural Science Foundation of China(Nos.21973094, 22173104, 22173105), and the Opening Project of PCOSS of Xiamen University (No.201908).We also thank Prof.Bin Jiang and Mr.Yaolong Zhang at University of Science and Technology of China for their technical help.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.03.080.
 Chinese Chemical Letters2022年11期
Chinese Chemical Letters2022年11期
- Chinese Chemical Letters的其它文章
- Distinct structural characteristics define a new subfamily of Mycoplasma ferritin
- Insight into the in vivo fate of intravenous herpetrione amorphous nanosuspensions by aggregation-caused quenching probes
- A TICS-fluorophore based probe for dual-color GSH imaging
- Ruthenium-modified porous NiCo2O4 nanosheets boost overall water splitting in alkaline solution
- Oral colon-targeted mucoadhesive micelles with enzyme-responsive controlled release of curcumin for ulcerative colitis therapy
- Fmoc-protected amino acids as luminescent and circularly polarized luminescence materials based on charge transfer interaction