
A general method for one-step synthesis of monofluoroiodane(III)reagents using silver difluoride
2022-12-07 08:26JingRenMengChengJiaFengHuanDuChiZhang
Chinese Chemical Letters 2022年11期
Jing Ren, Meng-Cheng Jia, Feng-Huan Du, Chi Zhang
State Key Laboratory of Elemento-Organic Chemistry, The Research Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China
Keywords:Monofluoroiodane(III) reagents Silver difluoride Iodobenzene derivatives One-step synthesis DFT calculations
ABSTRACT Herein we report a new general method for one-step synthesis of four kinds of fluoroiodane(III) reagents by treating the corresponding aryl iodides with silver difluoride (AgF2).This is the first method applicable for the synthesis of all four fluoroiodane(III) reagents including p-iodotoluene difluoride (1), fluorobenziodoxole (2), fluoro-benziodoxolone (3), and fluoro-N-acetylbenziodazole (4).AgF2 was firstly employed in the direct oxidative fluorination of iodobenzene and thus has shown its outstanding oxidation and fluorine-transfer ability.The use of AgF2 has improved the synthesis of fluoroiodane(III) reagents by shortening the reaction steps, avoiding the use of hazardous reagents, and simplifying the experimental operations.It was worth noting that we have developed the first one-step direct synthetic method for 3,while 3 can only be synthesized through Cl→F ligand exchange reaction previously.
Fluorine-containing organic compounds play increasingly important roles in pharmaceutical and agrochemical industry.Among the newly marketed pharmaceuticals every year, the proportion of fluoro-pharmaceuticals increased rapidly, which reached 50%in 2018 and 43% in 2019 [1].In agrochemical field, it is striking that fluoro-agrochemicals have accounted for 53% of all successful agrochemicals [2].Consequently, developing different methods for the introduction of fluorine atoms into organic molecules is of great significance.The growing demand of fluorination methods promoted the rapid development of fluorine-containing reagents, which are essential tools to introduce fluorine atoms.Among diverse reagents, hypervalent iodine(III) monofluorinetransfer reagents are becoming popular in recent years.
With the booming development of hypervalent iodine(III)reagents as multipurpose functional-group-transfer reagents [3–10], ones containing I-F bond, includingp-iodotoluene difluoride(p-TolIF2) (1), fluorobenziodoxole (2), fluorobenziodoxolone (3) and fluoro-N-acetylbenziodazole (4) (Fig.1), have received widespread attention in fluorine chemistry.They act as both a potent oxidant and a fluorine source in fluorination reactions.Various fluorination reactions achieved by reagents 1 and 2 have been reported,includingα-fluorination of carbonyl compounds [11], fluorinative rearrangements of styrenes and related compounds [12–14], ringopening 1,3-difluorination of cyclopropane [15] and oxyfluorination of diazocarbonyl compounds [16,17], various cyclization fluorination reactions [18–21], Balz–Schiemann fluorination [22]; it is worth noting that the rearrangement fluorinated products from some reactions mediated by 2 cannot be generated by N-F reagents[20,21], which demonstrates the unique reactivity of fluoroiodane(III) reagents.4 is the first monofluorine-transfer hypervalent iodine(III) reagent containing an I-N five-membered heterocycle,which was developed by our group in 2021.4 cannot only achieve intramolecular ring expansion fluorination of unactivated cyclopropanes, but also realize a series of known fluorination reactions[23].
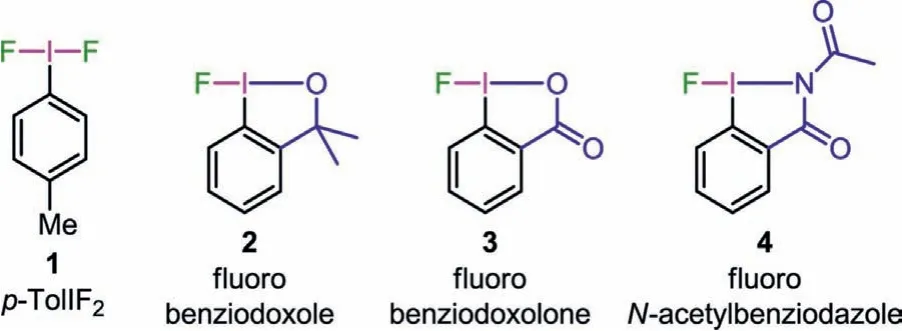
Fig.1 .Monofluoroiodane(III) reagents containing I-F bond.
Because of the versatility of fluoroiodane(III) reagents in fluorination reactions, it is of great significance to develop a simple and effective method to synthesize these reagents.Existing methods for their synthesis can be divided into two categories: ligand exchange reactions and one-step oxidative fluorination reactions (Scheme 1).Ligand exchange reactions are the most commonly used methods to synthesize reagents 1, 2 and 3 (Scheme 1a), which including: (1) 1 can be afforded through the ligand exchange reaction ofp-iodosyltoluene and aqueous HF [24,25] or the reaction ofpiodotoluene dichloride (p-TolICl2) and aqueous HF in the presence of HgO [26]; Togniet al.developed the TCICA/KF (spray-dried) system and applied it to the synthesis of iodane(III) difluorides in one pot in 2019 [27]; (2) 2 was often synthesized through Cl→F ligand exchange reactions [28]; ligand exchange reactions of OH→F,OTs→F, and OCOCF3→F are also efficient ways reported by Stuartet al.[29]; (3) 3 was synthesized firstly by Biber and Togniet al.through the reaction of its chloride precursor and spray-dried KF in 2016 [30].Although ligand exchange is the most commonly used method for the synthesis of these reagents, they are usually accomplished by multi-step synthesis, that is,p-iodosyltoluene,p-TolICl2, and cyclic iodane(III) precursors all need to be prepared in advance, and then ligand exchange is carried out with various organic or inorganic nucleophilic fluorine sources; but in most cases,HF together with ligand trapping additives, like the harmful HgO,are employed to remove the Cl ion.It is well known that aqueous HF is highly corrosive and HgO is highly toxic.As a common inorganic nucleophilic fluorine source in Cl→F ligand exchange reactions, KF absorbs moisture easily during use, which requires that the reactions must be carried out under strictly anhydrous and airfree conditions.
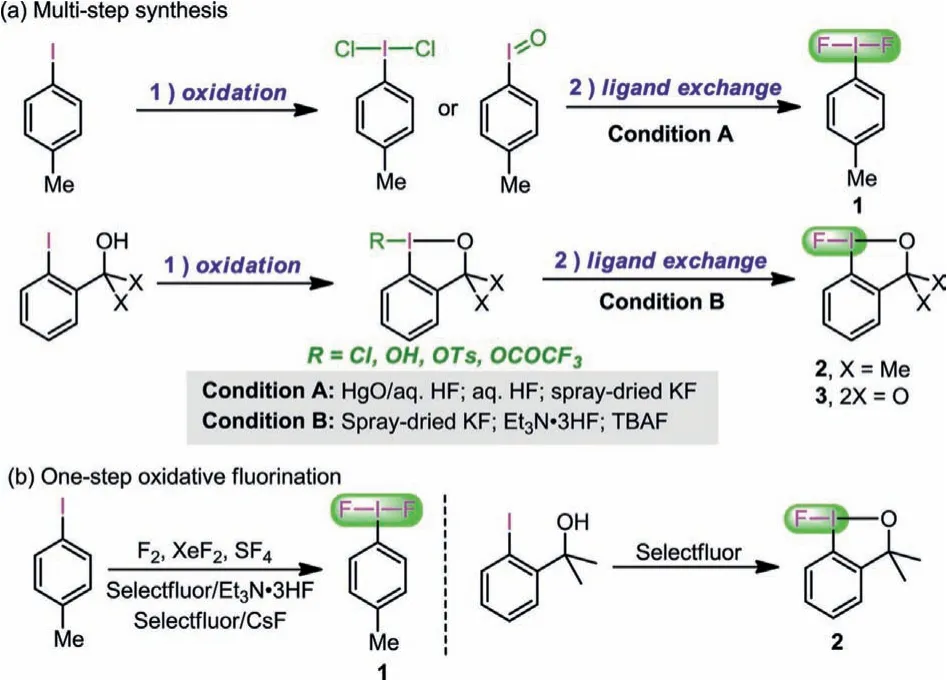
Scheme 1 .(a) Examples of multi-step synthesis of monofluoroiodane(III) reagents.(b) Examples of one-step synthesis of monofluoroiodane(III) reagents.
Reagents 1 and 2 can also be synthesized through one-step protocol (Scheme 1b).Fluorinating reagents with strong oxidative ability including F2[31,32], SF4[33] and XeF2[32,34] can directly oxidizep-iodotoluene to 1.However, F2and SF4are hazardous and highly toxic gasses at room temperature, causing experimental operations difficult in most laboratories.Direct electrosynthesis of 1 can be accomplished by anodic oxidation in the presence of Et3N·3HF or Et3N·5HF [35,36].Shreeveet al.[37] and Gilmouret al.[38] developed two systems of Selectfluor/Et3N·3HF and Selectfluor/CsF for one-step synthesis of 1.Prévostet al.reported that 2 can be directly synthesized by treating 2-(2-iodophenyl)propan-2-ol with Selectfluor [39].Unlike 1 and 2, the one-step method for 3 has not been developed, to the best of our knowledge.Although previous methods work well, there is not a direct general reagent for the oxidation of aryl iodides to the corresponding hypervalent iodine(III) monofluorine-transfer reagents of 1, 2, and 3.To synthesize these reagents more conveniently and efficiently,the development of a general one-step method is thus highly desirable.
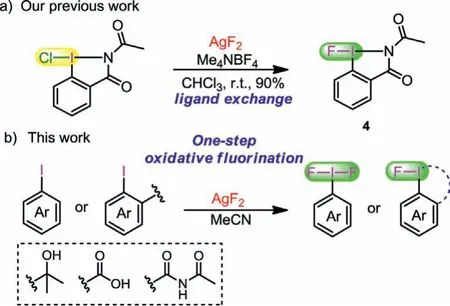
Scheme 2 .(a) Ligand exchange reaction of Cl→F mediated by AgF2.(b) One-step oxidative fluorination of aryl iodides.
Silver difluoride (AgF2), a commercially available fluorinating reagent with strong oxidation ability, has been used in few fluorination reactions.For example, Hartwiget al.applied it to the direct C-H fluorination of pyridines and diazines with exclusive siteselectivity [40].AgF2could also react with an excess of benzene in refluxing hexane to afford fluorobenzene [41].Recently, Tanget al.developed the ortho selective C-H trifluoromethoxylation of pyridine mediated by AgF2[42].In addition, AgF2can also act as a fluorine source in Cl→F ligand exchange reaction to synthesize fluoro-N-acetylbenziodazole (4) in high yield done by our group(Scheme 2a) [23].It is worth noting that AgF2(+1.98 V) [43] has stronger oxidation potential than Selectfluor (+0.33 V) [44].Therefore we speculated that AgF2would directly achieve the oxidative fluorination of the iodine center of aryl iodides, thereby providing a general method for the synthesis of fluoroiodane(III) reagents(Scheme 2b).
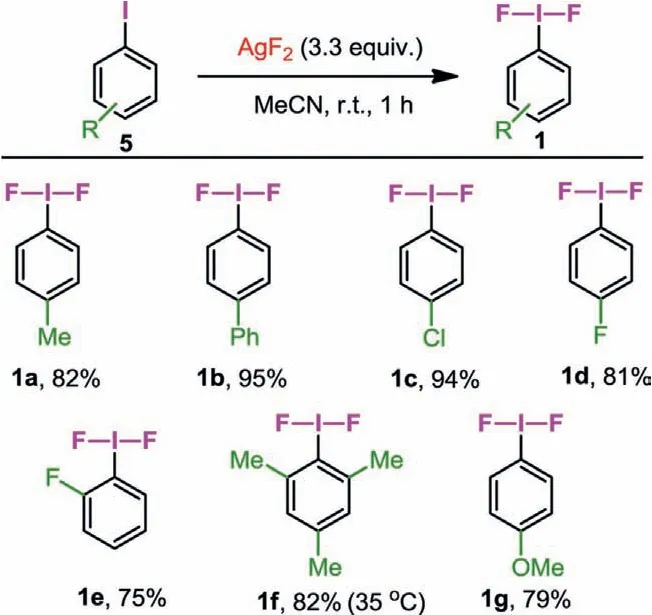
Scheme 3 .One-step synthesis of iodane(III) difluorides.Reaction conditions: Aryl iodide substrate 5 (1.0 mmol), AgF2 (3.3 mmol), 10 mL of anhydrous MeCN, r.t., 1 h,Ar atmosphere.Yields are for the isolated products.
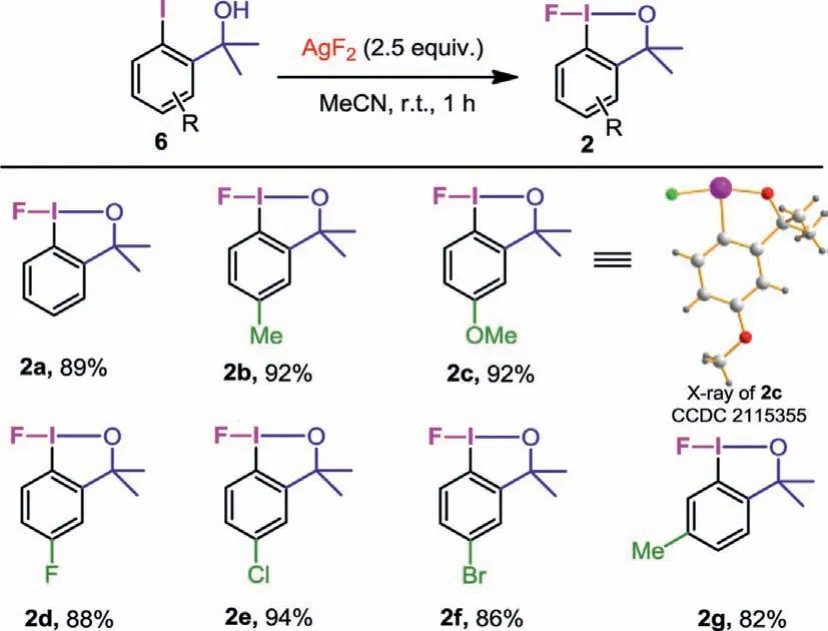
Scheme 4 .One-step synthesis of fluoro-benziodoxole.Reaction conditions: Aryl iodide substrate (1.0 mmol), AgF2 (2.5 mmol), anhydrous MeCN (10 mL), r.t., 1 h, Ar atmosphere.Yields are for the isolated products.
We began our preliminary investigation by trying to synthesize iodane(III) difluorides.4-Fluoroiodobenzene (5d) was allowed to react with 2.5 equiv.of AgF2in anhydrous acetonitrile at room temperature.We monitored the reaction through the1H NMR spectroscopy of the reaction mixture, and pleasantly observed two new signals other than 5d (Fig.2).However, only 77% of the raw material was converted, and prolonging the reaction time was ineffective.Therefore, we increased the amount of AgF2to 3.3 equiv.and found that 5d was converted completely after 1 h.The proton spectrum of the reaction mixture is quite clean in which no byproduct was observed.By referring to the1H NMR data of 1d reported previously [45], we concluded that H3and H4belong to 1d.After simple purification, we obtained the desired fluoroiodane(III)product 1d in 81% yield.
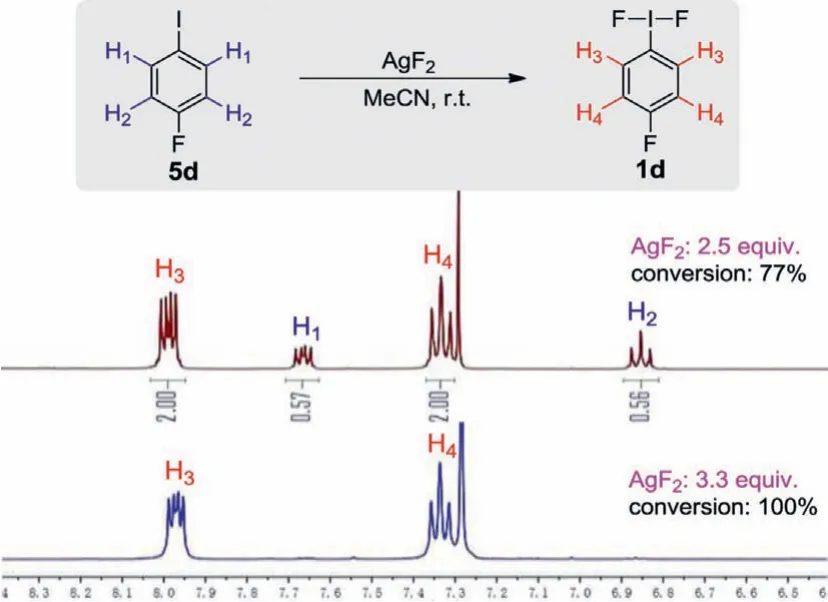
Fig.2 .1H NMR (CDCl3, 400 MHz) spectroscopy of the reaction mixture of 5d.
Subsequently, a series of iodane(III) difluorides 1a-1g containing various substituents were synthesized by this method (Scheme 3).In the presence of 3.3 equiv.of AgF2,p-TolIF2could be obtained in 82% yield within 1 h.The reaction of various aryl iodides bearing electron-withdrawing groups including phenyl (5b), chlorine (5c)and fluorine (5d) atoms proceeded smoothly, affording their corresponding iodane(III) difluorides 1b-1d in 81%–95% yield within 1 h.ortho-Fluoroiodobenzene (5e) and 2,4,6-trimethyliodobenzene (5f)could generate the corresponding desired products in good yields.Under the same condition, 1g was obtained in 79% yield.However,the stability of 1g is not as good as 1a-1f under ambient temperature, so we suggest that 1g should be stored in the freezer after work-up as soon as possible.
Encouraged by the exceptional performance of AgF2in the synthesis of iodane(III) difluorides, we turned our attention toward the application of AgF2in the one-step synthesis of cyclic fluoro-benziodoxole (2) (Scheme 4).By monitoring the reaction in the same way, we found that 2.5 equiv.of AgF2was enough to make 6 react completely within 1 h (see Supporting information for details).Under this simple condition, 2a was obtained in 89% yield.Electron-donating substituents on thepara-position of the iodine (methyl 6b and methoxy 6c) did not affect the reaction, the corresponding desired products 2b and 2c were obtained in 92% yield respectively.The structure of 2c was unambiguously confirmed by X-ray crystallography.A single crystal of 2c was grown in chloroform/n-hexane at room temperature, and X-ray crystallographic analysis of 2c showed that the F−I−O bond angle was 166° and the length of I-F bond was 2.0988 ˚A.Aryl iodides with electron-withdrawing substituents on thepara-position of the iodine (fluoro 6d, chloro 6e, bromo 6f) could also react smoothly, providing the desired products 2d, 2e and 2f in excellent yields.Significantly, reagents 2b-2f all have good stability and can be stored in the refrigerator for at least 3 months.2g bearing a methyl group on themeta-position, was obtained in 82% yield, but slight decomposition of it was observed after work-up.
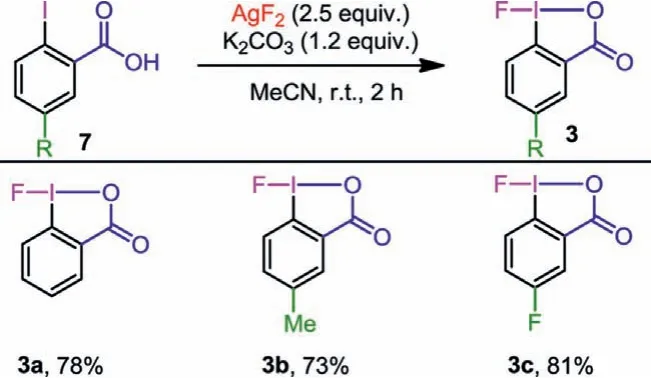
Scheme 5 .One-step synthesis of fluoro-benziodoxolone.Reaction conditions: Aryl iodide substrate (1.0 mmol), K2CO3 (1.2 mmol), AgF2 (2.5 mmol), anhydrous MeCN(10 mL), r.t., 2 h, Ar atmosphere.Yields are for the isolated products.
Fluoro-benziodoxolone (3), another important fluoroiodane(III)reagent, can only be prepared by the multi-step synthesis involving Cl→F ligand exchange reaction.Therefore, we are interested in exploring the one-step synthesis of 3 by AgF2.Initially, the reaction of 2-iodobenzoic (7a) and AgF2ind3-MeCN was performed and monitored by means of1H NMR spectroscopy.After 1 h, an aromatic main product can be observed on the spectrum (see Supporting information for details).According to the1H NMR data of 3a reported previously, there is no doubt that the new compound is our desired product.However, 3a was easy to decompose in the course of separation and purification.We speculated that HF, as a by-product of AgF2oxidative fluorination reaction of 7a, was very likely to form a hydrogen bond with the fluorine atom in 3a, thus activating 3a and making it easy to decompose [46–48].To verify our speculation, we added K2CO3(1.2 equiv.) to the reaction and the desired product 3a was isolated successfully as a white solid in a 78% yield without decomposition (Scheme 5).Replacing K2CO3withN,N-diisopropylethylamine gave inferior result.Consequently, we decided to add K2CO3as an additive to the reaction system.5-Methyl-and 5-fluoro-2-iodobenzoic acid (7b and 7c) also reacted smoothly and afforded the corresponding 3b, 3c in 73% and 81% yield successfully.Compared with the ligand exchange reaction mediated by spray-dried KF, our one-step protocol does not require a glove box during the reaction, which is convenient for researchers.
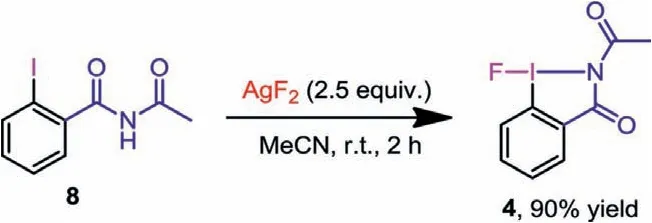
Scheme 6 .One-step synthesis of 4.
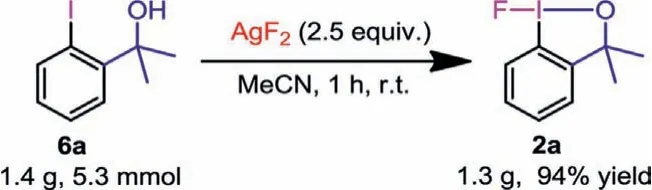
Scheme 7 .Large-scale synthesis of 2a.
We finally tried to synthesize fluoro-N-acetylbenziodazole (4)in one step.By treatingN-acetyl-2-iodobenzamide (8) with 2.5 equivalent of AgF2, we obtained 4 as a white solid in 90% yield(Scheme 6).4 is a new hypervalent iodane(III) monofluorinetransfer reagent containing a I-N five-membered heterocycle.The uniqueN-acetylbenziodazole skeleton would give it unique reactivity, and we have applied it to the intramolecular ring expansion fluorination of unactivated cyclopropanes [23].
To elucidate the mechanism of the direct oxidative fluorination of 6a by AgF2, we performed DFT calculation (Fig.3).We thought this reaction would feature a bimolecular radical oxidation pathway.Initially, AgF2forms a stable complex 1 with two acetonitrile molecules.The reaction starts with IM1, which is a stable complex generated from 6a and AgF2by coordination interaction.Subsequently, a concerted hydrogen-atom abstraction process occurs with a barrier of 6.2 kcal/mol, yielding the HF, AgF and IM2.Next,the fluorination process mediated by the second equivalent of AgF2would form 2a.The Gibbs free energy profile of the overall reaction pathway indicates that the fluorination process of iodine is the rate-limiting step, with an activation energy of 11.0 kcal/mol.The overall reaction pathway explains the use of excess equivalent of AgF2and the mild reaction conditions.In addition, this mechanism also provides a mechanism clue for the synthesis of other fluoroiodane(III) reagents by AgF2.
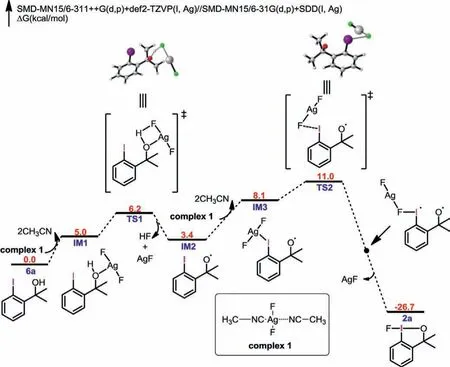
Fig.3 .DFT-computed potential energy profile for the reaction between AgF2 and 6a (Standard state: 25 °C, 1 mol/L).
We also performed a large-scale synthesis of 2a.When the reaction was scaled up to 5.3 mmol of 6a, the yield and reaction time were not affected at all (Scheme 7).
In conclusion, we have developed a general one-step method for rapid and efficient synthesis of the hypervalent iodane(III)monofluorine-transfer reagents from their corresponding aryl iodides by means of AgF2.Through this oxidative fluorination method, all four kinds of fluoroiodane(III) reagents are obtained.This method has the advantages of simple operation, high yield,step economy, and no use of harmful reagents.Noteworthy, we have developed the first one-step method for the synthesis of 3,and 3 can only be synthesized through Cl→F ligand exchange reaction previously.The convenient and rapid synthesis of monofluoroiodane(III) reagents would lay foundation for their widespread applications.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (Nos.21772096 and 22071116) and the National Key Research and Development Program of China (No.2017YFD020030202).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.01.070.
 Chinese Chemical Letters2022年11期
Chinese Chemical Letters2022年11期
- Chinese Chemical Letters的其它文章
- Zeolite-based Fenton-like catalysis for pollutant removal and reclamation from wastewater
- 1,n-Thiosulfonylation using thiosulfonates as dual functional reagents
- Degradation of florfenicol in a flow-through electro-Fenton system enhanced by wood-derived block carbon (WBC) cathode
- Simultaneous determination of indole metabolites of tryptophan in rat feces by chemical labeling assisted liquid chromatography-tandem mass spectrometry
- Self-powered anti-interference photoelectrochemical immunosensor based on Au/ZIS/CIS heterojunction photocathode with zwitterionic peptide anchoring
- The role of Cs dopants for improved activation of molecular oxygen and degradation of tetracycline over carbon nitride