
不对称单氟代、二氟甲基化反应研究进展
2022-12-06 12:42吴晶晶吴范宏
应用技术学报 2022年4期
郑 程, 吴晶晶, 吴范宏
(1.上海应用技术大学 化学与环境工程学院,上海 201418;2.上海绿色氟代制药工程技术研究中心,上海 201418)
1886年,Groult等[1]通过在金属铂容器中电解氟化钾和无水氢氟酸首次制备出氟气,开启了当代氟化学的研究进程。此后的一个多世纪,随着有机氟化学的不断发展,有机氟化合物已在医药、生命和材料科学等诸多学科领域得到了日益广泛的重视和应用[2-4]。将氟原子或含氟基团引入到有机分子中能显著地改变原有分子的物理化学特征,改善药物的亲脂性,提高生物利用度;通过影响化合物的构象,增强与靶标蛋白的结合能力,增加药物在靶组织的分布;通过阻断易代谢位点提高药物代谢稳定性,改变小分子的药代动力学性质等。药物设计中的氟基团取代策略成为药物结构修饰与改造的重要研究策略。2018-2020年,3年美国食品药品监督管理局(Food and Drug Administration)批准的118个新分子实体药物中,含氟小分子药物占被批准新药数目的38%,其中有26个手性含氟药物[5-7]。因此,如何在有机分子中高选择性地引入氟原子和含氟基团是有机化学、药物化学等领域的重要课题和研究热点。目前构建含C-CF、C-CF2键立体中心的化合物主要有两种策略,一种是通过亲核或亲电过程进行的不对称氟化反应,另一种是利用含有CF、CF2的砌块通过不对称氟烷基化反应得到手性单氟代、二氟烷基化小分子[8-11],目前已有综述对二氟甲基化与单氟甲基化的的反应进行了分析和总结[12-13],因此不再赘述,但对于不对称单氟代、二氟甲基化反应和总结鲜有报道,本文主要就近4年来的不对称单氟、二氟烷基化反应的研究进展进行简单的总结与展望。
1 不对称二氟烷基化反应
二氟烷基被认为是羟基与羰基的生物电子等排体,因此,将二氟烷基选择性地掺入生物活性分子中一直被认为是药物研发中的一种强有力的策略[14-15],这引起了化学家们极大的兴趣。
1.1 在环丙烷上进行C—CF2手性中心的构建
环丙烷环广泛存在于天然和合成生物活性分子以及材料科学中[16]。环丙烷环作为一个受限制的碳环,其引入可以显著改变靶标的生物学特性。,氟化环丙基化合物结合了氟原子和环丙烷的特征,成为受关注的分子骨架。尤其是在环丙烷环上构建一个含C-CF2键的立体中心将在药物合成中具有重要意义。比如对抗丙型肝炎病毒的伏西瑞韦等(见图1)。
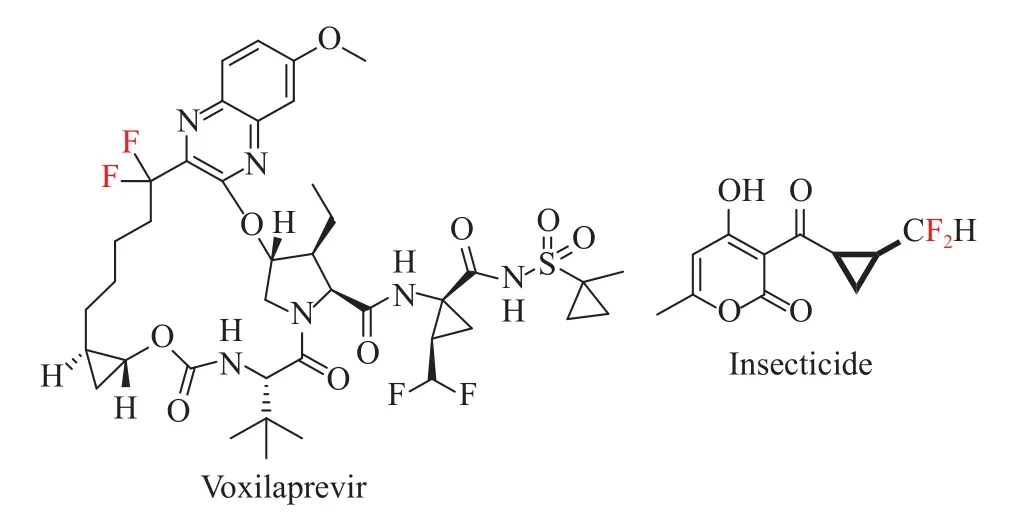
图1 含有二氟甲基环丙烷的药物分子Fig.1 Drug molecules containing difluoromethylcyclopropane
2017年,Duan等[17]开发了铁催化的环丙烷化反应,以硫叶立德作为二氟卡宾前体获得反式二氟甲基环丙烷,并且对于烯烃上有苯环取代以及带有吸电子或给电子苯环的取代基都具有出色的非对映选择性和高收率,但是对于脂肪族烯烃却表现出较低的反应性,且得到的产物具有较低的非对映选择性,见图2(a)。与此同时,Maxence等[18]使用了独特的铑催化剂首次报道了α-芳基重氮乙酸酯和α-硝基重氮酮的高官能化二氟甲基环丙烷不对称合成,在Maxence的研究中,他们使用了α-二氟甲基化烯烃作为二氟甲基化试剂,在催化剂Rh2((S)-BTPCP)4的催化下,高对映选择性地构建了含有C-CF2H键的四级碳立体中心,见图2(b)。该方法对于苯环骨架上带有给电子基团以及含卤素的重氮酮底物都可以获得优秀的产率(95%)以及优秀的对映选择性(99%),且对于底物中存在苯并呋喃环时也具有很好的兼容性,但是对硝基衍生物的产率很低。他们还测试了各种α-芳基重氮乙酸酯,发现在对位、邻位和间位甲基取代以及单个卤素取代的芳基重氮乙酸酯产率与ee值都较高,但是,当芳基上同时含有2个卤素以及以α-苯乙烯基重氮乙酸酯为底物时产率有所下降。这类反应的底物范围相当广泛,他们为这类用途广泛的化合物提供了一条新的合成途径。2018年,Cao等[19]报道了一种具有高非对映选择性和对映体选择性的环丙烷与相邻的具有单氟甲基(CH2F)基团的全碳四元立体中心的合成。无论重氮氧化合物上取代基的性质和位置如何,使用含氟烯烃的环丙烷化反应都很好地以57%~80%的收率和87%~95%的ee得到了具有α-CH2F基团的所需螺环丙烷,并且制备了具有邻近全碳立体中心的药物目标分子3-螺环丙基氧吲哚,此外,他们还对该反应体系进行了理论计算研究。研究结果表明,氟化溶剂与底物和反应中间体之间形成C-F···H-X相互作用可以极大地促进反应,见图2(c)。最近,Zhang等[20]通过用简单的方法合成了一种外消旋环丙基锌试剂,能够中高效地合成一系列有用的手性环丙烷,包括手性氟烷基化环丙烷和手性侧链对映体过量环丙烷。这一策略首先是镍催化剂与烷基亲电试剂的对映体选择性偶联,紧接着将剩余的单一构型的环丙基锌试剂与各种亲电体进行立体专一性偶联,在环丙烷环上制备2种构型相反的官能团化手性环丙烷。这些手性环丙烷能够作为不同转化反应的多功能合成砌块,从而获得具有药用价值的结构多样化的分子,见图2(d)。
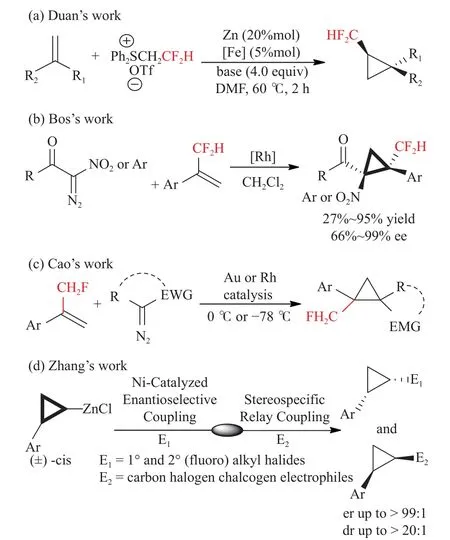
图2 环丙烷结构中的不对称二氟甲基化反应[17-20]Fig.2 Asymmetric difluoromethylation in cyclopropanestructure[17-20]
1.2 杂环、吲哚及其衍生物不对称二氟烷基化反应
许多具有良好的药理活性的药物分子中都存在有杂环、吲哚结构[21]。因此,开发简便的吲哚对映体选择性的合成方法已成为有机合成化学领域的研究热点[22-27]。在目标有机分子中选择性地掺入氟烷基往往会显著改变分子的代谢稳定性、生物利用度、亲脂性、膜透性等性质,在药学研究中有着广泛的应用[28-30]。因此,将氟代烷基团选择性地引入到目标有机分子中具有重大意义。
2017年,Li等[31]报道了二氟烯氧基硅烷在手性磷酸催化下参与的不对称二氟甲基化反应,高对映选择性地合成了各种二氟烷基化吲哚-3-酮类化合物(见图3),此外,通过对产物进行简单的转化,产物可以很容易地生成为旋光性酯类和α-CF2H衍生物(见图4)。经过研究发现,手性磷酸催化剂在反应体系中发挥着至关重要的作用,并提出了一个反应中可能存在的潜在的过渡态,磷酸催化剂通过氢键作用激活环酮亚胺,从而创造了烯醇硅醚优先攻击C=N的下方的手性环境,关于Mukaiyama-Mannich反应的详细机理他们还在进一步的研究(见图5)。
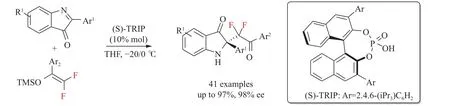
图3 不对称二氟烷基化吲哚-3-酮类衍生物的合成策略[31]Fig.3 Synthesis strategy of asymmetric difluoroalkylated indole-3-one derivatives[31]
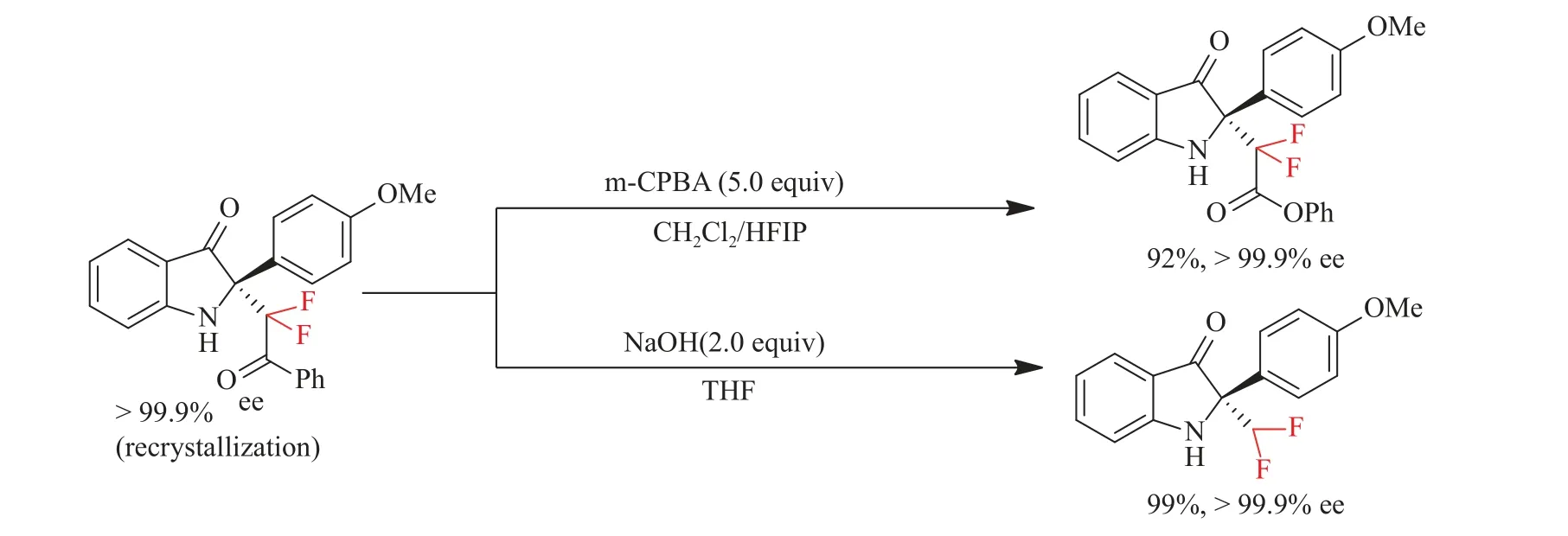
图4 吲哚-3-酮类衍生物的进一步转化[31]Fig.4 Further transformation of indole-3-one derivatives[31]
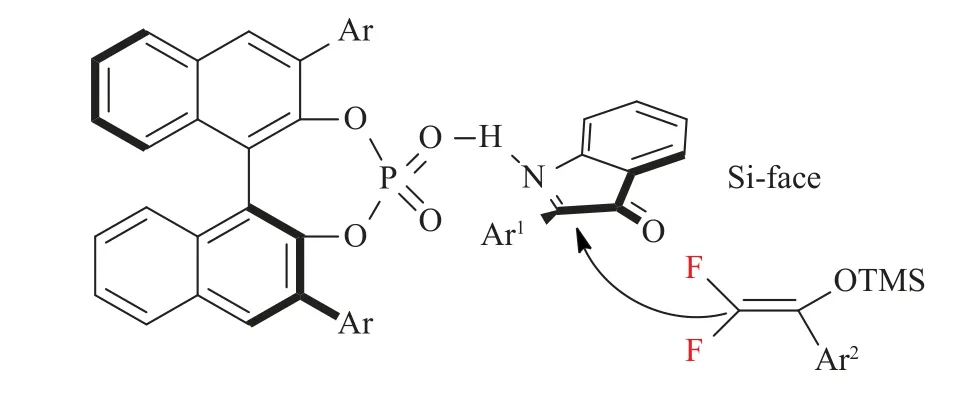
图5 可能的过渡态结构[31]Fig.5 Proposed transition state structure[31]
2017年,Lin等[32]报道了以商品化氟烷基磺酰氯为自由基源,在Cu(I)/CPA催化的烯烃与二氟甲基磺酰氯的不对称自由基分子内氨氟烷基化反应,并且在苯环上具有不同取代基(R1)的N-芳基脲底物中进行探索,他们发现,无论是芳环上不同位置存在吸电子基团或给电子基团或是带有卤素,反应均平稳进行,得到相应的产物产率均较高。ee值为92%~95%。并且对于具有取代基(R3)的底物也适用于通用的反应条件,以中等产率,以及较高的ee值获得产物,见图6(a),他们还探索了通过使用其他氟烷基磺酰氯来替换二氟甲基磺酰氯在0℃反应条件下也能使反应顺利进行,见图6(b),并且他们利用合成的产物进行了进一步的转化,合成了天然生物碱吡咯里西啶的衍生物(见图7)。
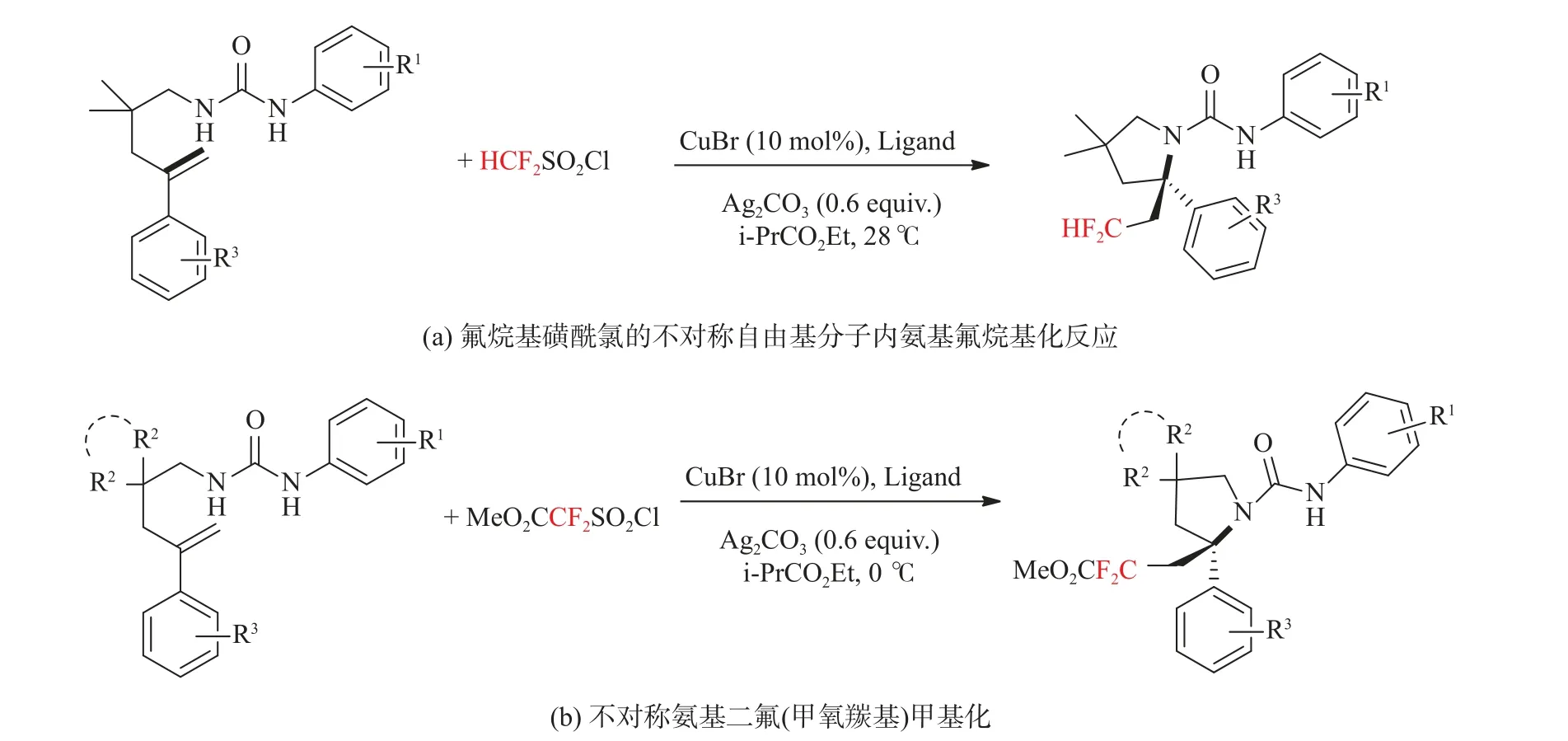
图6 不对称自由基分子内氨基二氟甲基化反应[32]Fig.6 Intramolecular aminodifluoromethylation of asymmetric radicals[32]
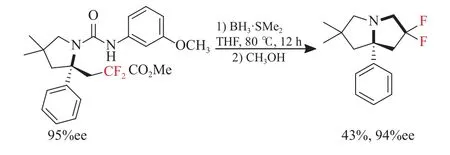
图7 吡咯里西啶衍生物的合成[32]Fig.7 Synthesis of pyrrolidine derivative[32]
取代的4H-色烯酮是化合物中常见的重要结构,具有广泛而重要的生物活性,特别是手性色烯酮具有优异的抗炎、抗氧化和抗菌活性(见图8)。2018年,Wang等[33]报道了一种通过一锅法卡宾催化的聚合/氧化过程合成对映体过量的氟甲基取代色满酮的新策略(见图9)。首先卡宾催化剂与醛形成Breslow中间体Ⅰ后,紧接着缺乏电子的双键发生分子内质子转移,得到氢酰化中间体Ⅱ,再通过释放卡宾与烷基结合,最后被氧化得到含氟甲基取代的色烯酮。在这个过程中,二甲基亚砜中催化量的碘被用作氧化剂,此外,整个反应过程中没有涉及到金属催化,避免了金属残留的问题。
图8 各种手性色烯酮[32]Fig. 8 Variouschiral chromophenones[32]
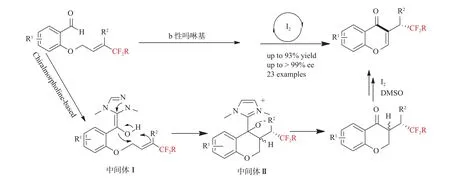
图9 卡宾催化的合成对映体过量的氟甲基取代色满酮的新策略[33]Fig.9 A new strategy for carbene-catalyzed synthesis of enantiomeric excessfluoromethyl substituted chromanones[33]
1.3 羰基化合物的不对称二氟烷基化反应
2018年,Liu等[34]以碘二氟乙酸乙酯为自由基源,以高达67%的收率和95∶5的er值提供了含Rf的化合物,成功地实现了β-酮酯的自由基的二氟烷基化,该反应对芳环上含有供电子基或吸电子基的酮酯底物均有良好的兼容性,(见图10)该研究揭示了对映体选择性自由基二氟烷基化的光氧化还原催化途径。首先,用可见光激发光催化剂A,光催化剂B的激发态可被配合物D还原猝灭,得到配合物E和Ir(II)。随后茚酮底物被手性刘易斯酸活化生成含镍烯酸的中间体F,并被自由基G捕获生成不稳定的中间体H。最后,[Ni/L1]在光照下变成激发态B或氧化态E,重新生成Ni(II)物种并释放出最终产物(见图11)。
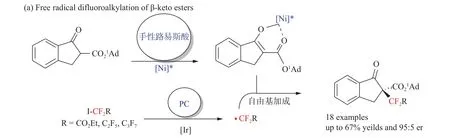
图10 碘二氟乙酸乙酯对β-酮酯的不对称二氟甲基化反应[34]Fig.10 Asymmetric difluoromethylation of β-keto estersby ethyl iododifluoroacetate[34]
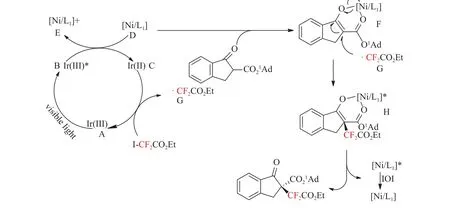
图11 可能的反应机理[34]Fig.11 Proposed reaction mechanism[34]
2019年,Endo等[35]报道了在手性亚磷酰胺-铜催化剂作用下,二氟碘甲烷和二乙基锌与吡啶等共溶剂通过锌/碘交换反应,以较高的产率和较高的对映选择性得到了二氟甲基化的Michael加成产物(见图12)。
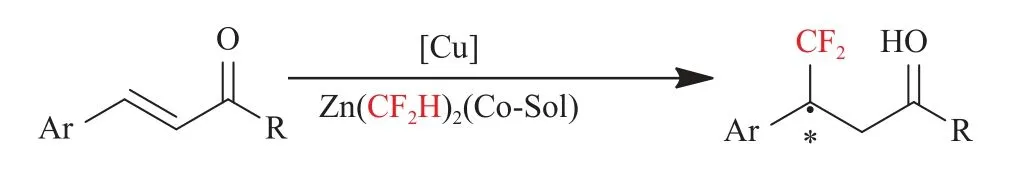
图12 (二氟甲基)锌试剂催化α,β-不饱和酸的不对称二氟甲基化反应[35]Fig.12 The catalytic enantioselective difluoromethylation of α,β-unsaturated acids with(difluoromethyl)zinc reagent[35]
2018年,Yu等[36]实现了Ni(II)催化的2-乙酰基氮烯与β-二氟甲基取代硝基烯烃的Michael不对称加成反应,以高达97%的收率和98%ee的对映选择性合成一系列带有季碳立体中心的新型二氟烷基化的吲哚酮类化合物。他们的研究显示硝基烯苯基环上取代基的电子性质对反应的对映选择性和反应活性影响不大。但是,硝基烯苯的苯环上的邻位取代基对反应有显著的不利影响,未能与苯环上带有邻甲氧基或邻氯基的底物发生反应,这可能归因于苯环邻位上的大的空间位阻,且对于庚基取代硝基烯,对映选择性较差(见图13)。此外,他们还进行了克级制备。最后他们提出了一个合理的不对称诱导过程。配体L1与Ni(acac)2配位形成镍配合物。随后,2-乙酰基氮芳烃与该镍配合物相互作用生成了烯醇酸。同时,硝基烯烃也通过与Ni配位而被活化。由于硝基烯烃的NO2基团与恶唑啉环的苯基之间的空间位阻,不利于烯酸酯在C=C平面上方进攻,导致主要在其反面进行加成(见图14)。
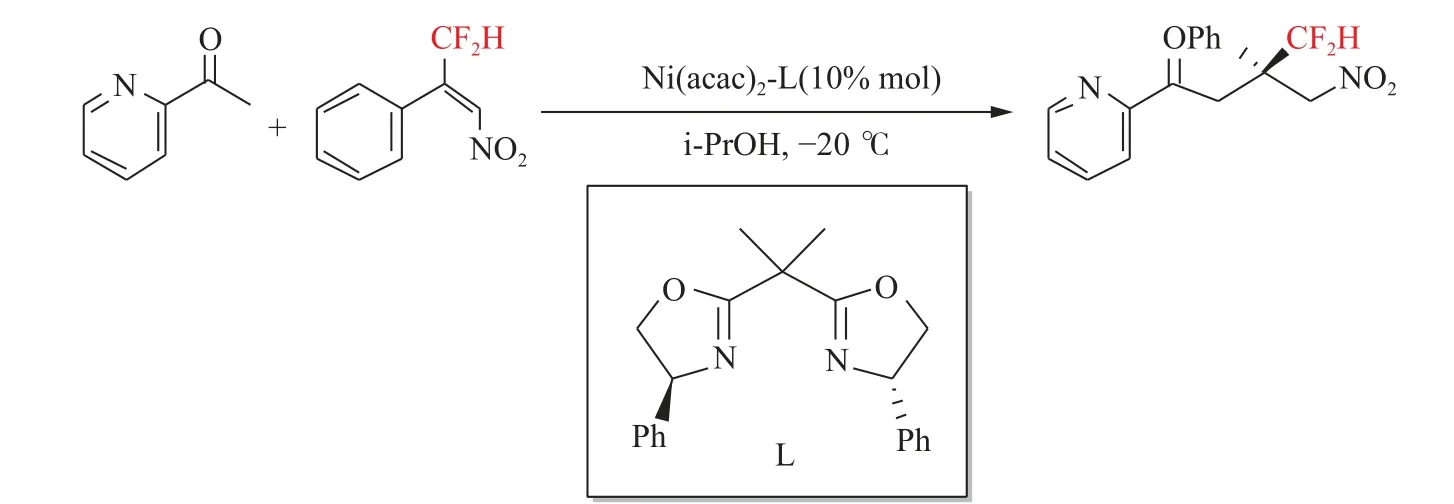
图13 2-乙酰基氮烯与β-二氟甲基取代硝基烯烃的不对称Michael加成反应[36]Fig.13 Asymmetric Michael addition reaction of 2-acetylazene withβ-difluoromethyl substituted nitroolefins[36]
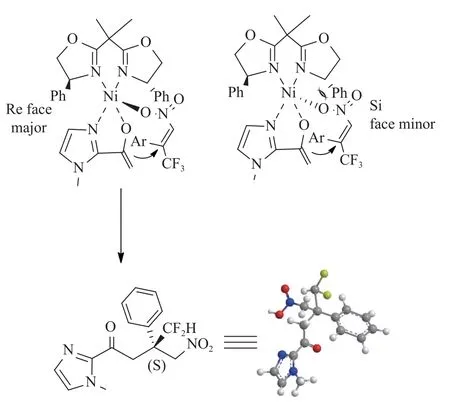
图14 可能的反应机理[36]Fig.14 Proposed reaction mechanism[36]
2019年,Liang等[37]报道了可见光氧化还原催化和手性路易斯酸催化相结合来构建一系列含氟的γ-酮酸衍生物(见图15)。采用这种双重催化策略,成功地合成了一系列含偕二氟烷基的手性γ-酮酰胺和一系列含氟的α,β-不饱和γ-酮酸酯,并具有较高的立体选择性。他们通过实验研究发现,除Lewis酸催化剂外,该工艺的化学选择性高度依赖于氟试剂。这种方法便于快速获得一系列重要的医药、增塑剂和各种其他添加剂的前体。
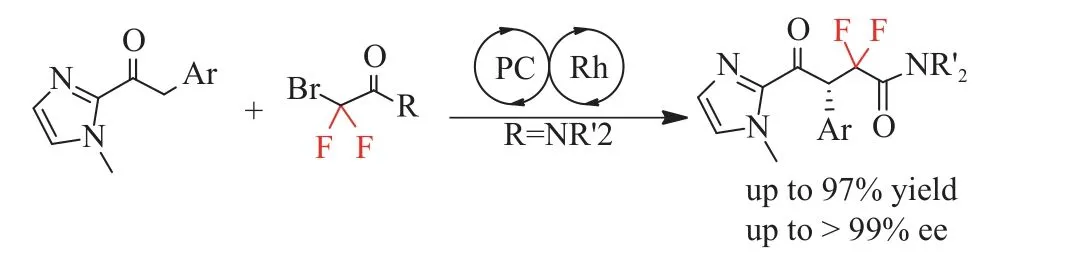
图15 含偕-二氟烷基的手性γ-酮酰胺的合成[37]Fig.15 Synthesis of chiral γ-ketoamides containing gemdifluoroalkyl[37]
1.4 炔烃、烯烃类化合物的不对称二氟烷基化反应
到目前为止,尽管许多集中在羰基化合物上的不对氟称烷基化反应被报道[38],但它们都局限于羰基化合物的官能团化。而碳碳三键的多种合成用途可以应用于生物活性分子和先进功能材料的合成,因此,含有C-CF2手性中心的炔烃类化合物可以作为合成多种有机化合物的通用砌块,从而为在药物化学和材料科学中的应用提供了一个有用的方案。
2018年,Gao等[39]首次报道了在铜催化下,以TMSCF2CONEt2作为氟化试剂直接与炔丙基磺酸盐进行不对称二氟甲基化反应,在最佳反应条件下,多种炔丙基磺酸盐顺利地进行了二氟烷基化反应,得到了具有较高ee值和立体选择性的产物,这些二氟烷基化炔烃通过常规的合成方法很难得到。此外,该反应还对多种官能团具有良好的兼容性以及较高的区域选择性,见图16(a)。所有的具有对映体的二氟烷基化炔烃的结构都是全新的,可以作为面向各种类型有机合成的通用砌块。接着在2019年,Gao等[40]报道了铜催化的二氟炔丙基磺酸盐在二氟烯氧基硅烷催化下的高对映选择性二氟烷基化反应。该反应以Cu(I)/PyBox为催化剂,在温和的反应条件下进行,见图16(b)。该方法具有产率高、对映体选择性高(对映体比可达99∶1)、官能团耐受性好等优点。由此得到的手性炔丙基α,α-二氟酮可以转化为对映体过量的炔丙基二氟甲基化化合物,在药物化学和材料科学领域中为这些有意义的手性二氟甲基化合物提供了一个很好的合成平台。
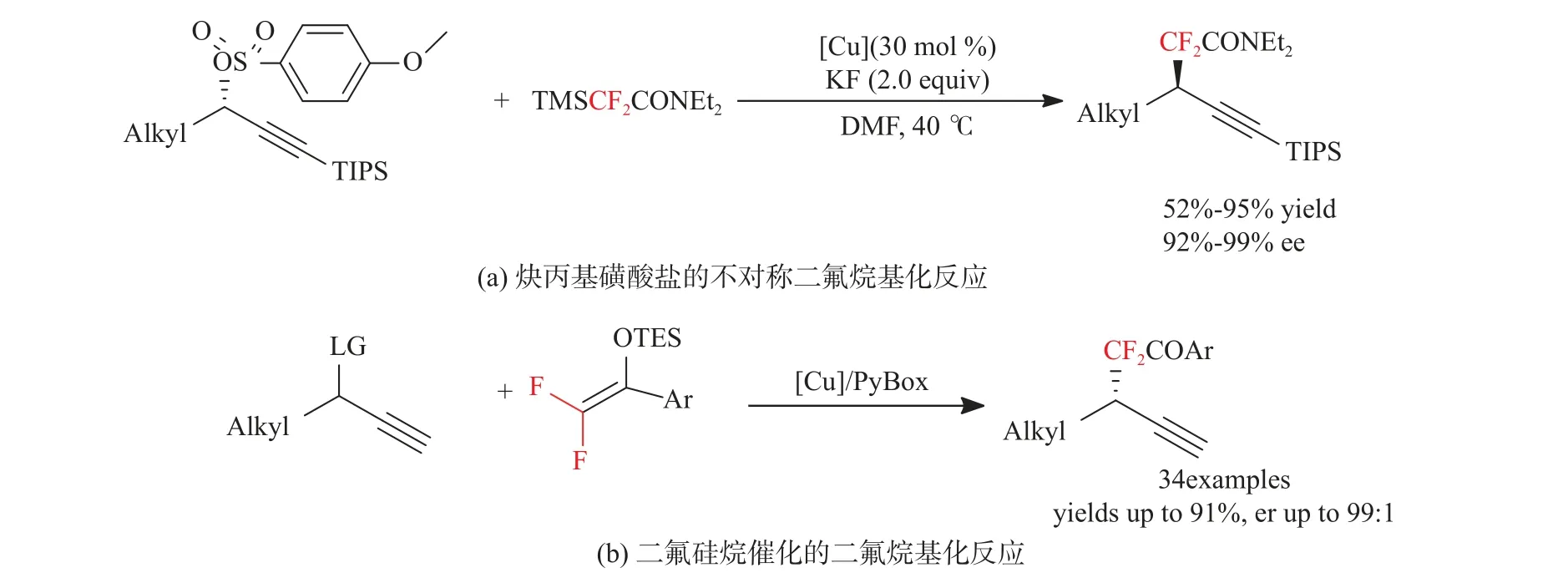
图16 炔基类化合物的不对称二氟烷基化反应[39-40]Fig.16 Asymmetric difluoroalkylation of alkynyl compounds[39-40]
2019年,Zhang等[41]首次报道了一种高效的Rh(II)催化的二氟重氮乙烷试剂(PhSO2CF2CHN2,Ps-DFA)催化的非末端炔烃的环丙烯反应(见图17)。这种不对称转化提供了广泛的对映体过量的二氟甲基化环丙烯的有效途径(40种底物,高达99%的收率,97%的ee值)。通过随后的立体变换过程,包括交叉偶合、加氢、Diels-Alder反应和Pauson-Khandd反应,证明了所获得三元环产物的合成用途。
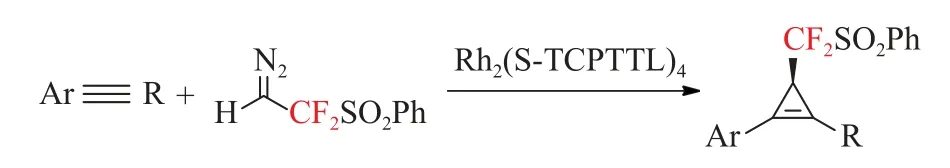
图17 非末端炔烃与重氮化合物的催化对映选择性环丙烯反应[41]Fig.17 Catalytic enantioselective cyclopropene reaction of nonterminal alkyne with diazo compounds[41]
2019年,Guo[42]报道了可见光诱导的Cu催化的烯烃的不对称腈基二氟烷基反应(见图18)。在该反应中,CuI起双重作用,起着传递电子的作用和作为选择性形成C-CN键的不对称交联催化剂。紧接着在2020年,ISRAR等[43]报道了烯烃在铜催化下的不对称氰基二氟烷基化反应(见图19)。从易得的化学品中获得一系列的手性氟代烷基氰化物,并且立体选择性优异。该方法使用氟代烷基碘作为氟代烷基源,反应条件温和。过氧化物LPO在促进该自由基不对称反应中起着重要作用。

图18 光诱导的Cu催化的烯烃的不对称腈基二氟烷基反应[42]Fig.18 The visible-light-induced Cu-catalyzed photoredox enantioselective cyanofluoroalkylation of alkenes[42]

图19 烯烃的不对称氰基二氟烷基化反应[43]Fig.19 Asymmetric cyanodifluoroalkylation of olefins[43]
2 不对称单氟烷基化反应
单氟烷基化合物相对于全氟烷基/三氟甲基化合物不仅在分子中可以引入一个氟原子还可以引入其他有机官能团,同时单氟亚甲基也具有改变相邻基团的酸碱性从而影响化合物的生物利用度,而且在合成步骤与经济上比全氟烷基化合物更具有优势[44],但目前已有的报道还很少。环丙烷化的C-F手性中心的构建:2019年,Amandine等[45]报道了Rh2((S)-TCPTTL)4介导的以α氟丙烯酸酯为原料催化不对称合成功能化的含氟环丙烷,以较好的产率、优良的dr和ee值得到了所需的含氟环丙烷。他们同时还对该反应体系进行了理论计算,通过计算总的翻转频率,对不同的α-芳基重氮乙酸酯的反应性差异的结果进行了合理的解释(见图20)。
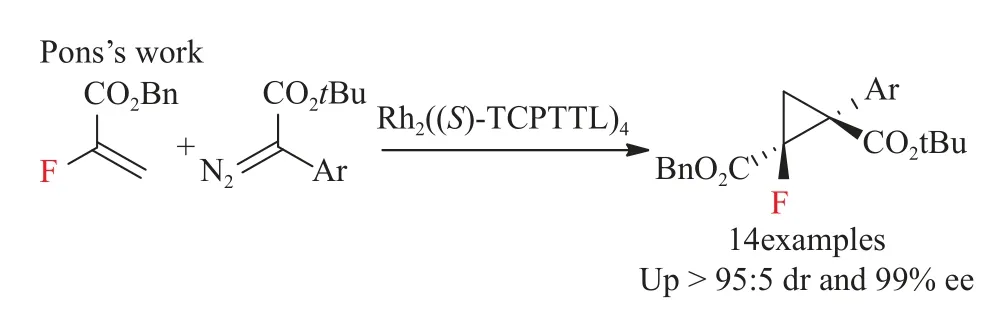
图20 环丙烷化的不对称单氟烷基化反应[45]Fig.20 Asymmetric monofluoroalkylation of cyclopropanation[45]
3 结语
从以上总结中可以发现,不对称单氟代、二氟甲基化反应在过去的4年间得到了一些发展。目前主要集中在以炔烃、烯烃、含羰基类、醇类化合物为反应原料,通过不对称氟化反应构建结构多样化的含氟的手性化合物,大部分反应类型为亲核、亲电取代反应、自由基反应、Michael加成等反应类型。
尽管过去4年中,不对称单氟代、二氟甲基化反应取得了重要的进展,但是相对而言,该领域还处于发展的起步阶段,已有的报道中反应模式还比较少,所采取的策略也比较单一。同时,该领域中还存在着较多的挑战:例如利用亲电或其它单氟、二氟烷基化试剂发展普通羰基α位的不对称单氟、二氟烷基化反应;用亲核或其他单氟、二氟烷基化试剂通过金属催化的偶联反应或者其他非金属催化的反应途径实现有机化合物的不对称单氟、二氟烷基化;在手性配体的作用下利用单氟、二氟烷基化试剂对烯烃直接进行不对称加成反应合成各种各样含有C-F、C-CF2键的立体中心的复杂有机化合物等。总而言之,有机化合物的不对称单氟代、二氟甲基化反应还存在着较大的发展空间。
猜你喜欢
分子催化(2022年1期)2022-11-02
石油石化绿色低碳(2019年6期)2019-01-14
石油石化绿色低碳(2018年5期)2018-03-20
国外医药(抗生素分册)(2016年4期)2016-07-12
国外医药(抗生素分册)(2016年2期)2016-07-12
石油炼制与化工(2016年6期)2016-04-06
设备管理与维修(2016年6期)2016-03-16
合成化学(2015年2期)2016-01-17
石油炼制与化工(2014年9期)2014-04-06
食品工业科技(2014年13期)2014-03-11
