
乌鳢水泡病毒核蛋白和磷蛋白多克隆抗体的制备与应用
2022-12-06 01:37张彦冰郭梦雅刘晓丹
水产学杂志 2022年5期
张彦冰,郭梦雅,刘晓丹
(扬州大学动物科学与技术学院,江苏 扬州 225000)
乌鳢水泡病毒(Snakehead Vesiculovirus,SHVV)属弹状病毒科(Rhabdoviridae)成员,其基因组为单股不分节段的负链RNA[1],是引起乌鳢(Channa argus)病毒病的主要病原[2]。感染乌鳢水泡病毒的鱼体多体表发黑,鳃无血色苍白,不摄食,游泳无力偶尔会狂躁不安,容易在水流速度慢的地方聚集;解剖可观察到病鱼的肝脾肾肿大,有腹水[3]。乌鳢水泡病毒的基因组中有编码核蛋白(Nucleoprotein,N)、磷酸化蛋白(Phosphoprotein,P)、基质蛋白(Matrix protein,M)、糖蛋白(Glycoprotein,G)以及RNA 依赖的RNA 聚合酶蛋白(RNA-dependent RNA polymerase,L)五个结构蛋白[3,4]。其中,核蛋白N 是含量最丰富、结构最保守的病毒结构蛋白,核蛋白与病毒基因组RNA 相结合,作为病毒转录与复制的模板,对转录和复制过程起着重要的调节作用[5]。与此同时核蛋白能够激活鱼体内的T 淋巴细胞,对病毒粒子产生免疫应答,也可以激活T 辅助淋巴细胞,一定程度上加强B 淋巴细胞生成抗体的能力[6]。磷酸化蛋白P 是促使基因组从转录状态转换到复制状态的关键因子,通过高度磷酸化改变自身结构,启动核衣壳向病毒基因组RNA 结合,在RNA 与蛋白质组成复合体的过程中发挥重要作用[7,8]。N 蛋白、P 蛋白与L 蛋白形成核糖核蛋白复合体指导病毒RNA 的转录与复制。
大肠杆菌(Escherichia coli)表达系统遗传学背景清晰、基因表达调控机理研究成熟,操作过程简便,能够快速诱导表达目的蛋白,且表达产量高,最为成熟、应用最普遍的原核表达系统,有多种适用不同蛋白的表达载体和宿主菌[9,10]。大肠杆菌表达系统缺乏磷酸化、糖基化等翻译后蛋白修饰系统,适合翻译后修饰作用不影响蛋白活性及特定结构的外源蛋白表达[11]。
本研究通过克隆乌鳢水泡病毒SHVV 的N、P蛋白基因,构建重组表达载体质粒pGEX-5x-1-N和pGEX-5x-1-P,利用大肠杆菌表达系统对N、P蛋白进行原核表达,纯化回收重组蛋白后免疫新西兰大白兔和小鼠获得N、P 蛋白的多克隆抗体,为该病毒免疫学检测方法的建立及疫苗的研发提供基础资料,并利用获得的抗体检测病毒蛋白在宿主细胞内的分布。
1 材料与方法
1.1 菌株、载体及细胞
大肠杆菌DH5α 与BL21(DE3)感受态细胞购于宝生物工程(大连)有限公司;实验中表达载体pGEX-5x-1 及SSN-1 细胞由本实验室保存;SHVV由本实验室分离并保存。
1.2 主要试剂
MEM细胞基础培养液、青霉素-链霉素溶液购自Hylcone 公司;胎牛血清(FBS)、胰酶购自Gibco 公司;限制性内切酶Sal I、EcoR I、Not I 和RNA提取与反转录试剂盒、T4 DNA ligase、2×Taq PCR Mix 购自宝生物工程(大连)有限公司;包被液、洗涤液、稀释液、底物液、终止液等购于武汉科前动物生物制品有限责任公司;质粒抽提试剂盒、PCR 产物纯化回收试剂盒等均购于北京康为世纪生物技术有限公司;SDS-PAGE 凝胶配置试剂盒与蛋白微量回收试剂盒购自于生工生物工程(上海)股份有限公司;其他试剂均为国产分析纯。
1.3 引物设计与合成
以SHVV N、P 序列为模板分别设计引物。N 基因上游引物命名NF-Sal I,序列为GGGTCGACTCATGGAAAACCAAATCATCAAG;下游引物命名NB-Not I,序列为GATGCGGCCGCTCACAAAGCTTG GTGCTTCAG。P 基因上游引物命名PF-EcoR I,序列为AGCGAATTCATGGCAAAACCAGTTTTCCA;下游引物命名为PB-Not I,序列为ACTGCGGCCGCTTAGAACAGCACCATTTGCT。
1.4 目的基因的扩增与纯化
用Trizol 法提取感染病毒细胞的总RNA,再根据反转录试剂盒说明书反转录成cDNA。以反转录的cDNA 为模板,用1.3 中所设计的特异性引物进行PCR 扩增。反应条件为:95℃预变性5 min;95℃变性30 s,55℃退火30 s,72℃延伸30 s,共30 个循环;延伸反应72℃5 min,4℃保存。PCR 产物经1%的琼脂糖凝胶电泳鉴定后,纯化回收目的片段。
1.5 重组表达质粒的构建及鉴定
用对应的限制性核酸内切酶将PCR 产物以及pGEX-5x-1 表达载体进行双酶切处理后,利用T4 DNA 连接酶将酶切后的目的片段与克隆载体连接,构建重组质粒,转入大肠杆菌DH5α 感受态细胞。将转化后的感受态细胞涂布于含有氨苄青霉素的LB 固体培养基平板上,于37℃培养箱培养12 h,挑取单菌落重新接种在LB 培养基上过夜培养,用质粒提取试剂盒提取质粒,并分别用Sal I/Not I 和EcoR I/Not I 对所提质粒进行双酶切鉴定,将鉴定结果为阳性的克隆且测序正确的重组表达质粒分别命名为pGEX-5x-1-N 和pGEX-5x-1-P。
1.6 重组蛋白的原核表达
将上述获得的质粒转化感受态细胞BL21(DE3)涂布LB 平板,挑取单克隆,37℃震荡培养。在OD 值达到0.6 左右时加入IPTG,28℃诱导6 h。诱导结束后,取2 mL 诱导菌液及对照菌液4℃5 000 r/min 离心5 min,收集菌体。用100 μL PBS 重悬菌体,加入等量的SDS-PAGE 上样缓冲液,沸水中处理10 min,取10 μL 样品进行SDS-PAGE 分析。
1.7 动物的免疫
利用蛋白凝胶回收试剂盒回收经SDS-PAGE分离的目的蛋白后,选取新西兰大白兔,初次免疫时加等量完全弗氏佐剂乳化,后两次加等量不完全弗氏佐剂,皮下多点注射500 μg 的pGEX-5x-1-N乳化后的蛋白,每隔两周免疫一次,共进行三次免疫;同样,选取4~6 周龄小鼠,每次免疫100μg 的pGEX-5x-1-P 乳化后的蛋白,免疫三次;设置未免疫的兔子和小鼠作为对照。
1.8 效价测定及Western-blot 检测
免疫结束后采血,装入离心管中,置于4℃过夜,取出析出的血清,分装备用,在获得抗血清之后用ELISA 测定血清效价。具体操作方法如下:用包被液(pH9.6,0.05 mol/L 碳酸盐缓冲液)稀释纯化的SHVV 病毒悬液抗原后,按照0.625 μg/孔的浓度包被聚丙乙烯酶标板,4℃过夜;弃去包被液,然后每孔加入200 μL 洗涤液(含0.05%Tween20 的0.01 mol/L、pH7.4 的PBS,PBST)洗涤4 次,每次5 min,在干毛巾上拍干;每孔加入100 μL 封闭液(含1%BSA 的PBS),37℃温育1 h,拍干,洗板同前;加入100 μL 适当稀释的多克隆抗血清(按1∶200~1∶25 600 倍比稀释),对照组加入正常兔子血清(按1∶200~1∶25 600 倍比稀释作为阴性对照),37℃温育30 min,洗涤同前;每孔加入100 μL 适当稀释倍数的辣根过氧化物酶标记的羊抗小鼠IgG 抗体,37℃温育30 min,同上洗涤;加底物液100 μL(底物液A 50 μL,底物液B 50 μL)显色10 min,每孔加终止液50 μL,于酶标仪630 nm 下测定光密度。再用0.1 MOI SHVV 感染SSN-1 细胞24 h 后收集细胞样品,Western blot 分析多克隆抗体的特异性。
1.9 间接免疫荧光检测
将SSN-1 细胞接种在24 孔细胞培养板中,待细胞长满单层后感染0.1 MOI(Multiplicity of infection,MOI)SHVV,同时设立未感染病毒的细胞作为对照组。接毒的细胞在28℃细胞培养箱培养24 h后,用预冷的丙酮-甲醇(1∶1)固定液固定10 min,PBS 清洗3 次后,1%BSA 封闭30 min,制备的N、P 蛋白的多克隆抗体(1∶5 000)为一抗,FITC 标记的商品化抗体(1∶200)为二抗,DAPI 细胞核染色3~5 min,最后在荧光显微镜下观察并拍照。
2 结果与分析
2.1 目的基因的扩增
以感染病毒的SSN-1 细胞cDNA 为模板,采取RT-PCR 方法通过特异性引物扩增出SHVV 相应的P、N 基因片段。琼脂糖凝胶电泳检测可见,对应片段大小分别为864 bp 和1 290 bp 的片段(图1),与预期结果一致。
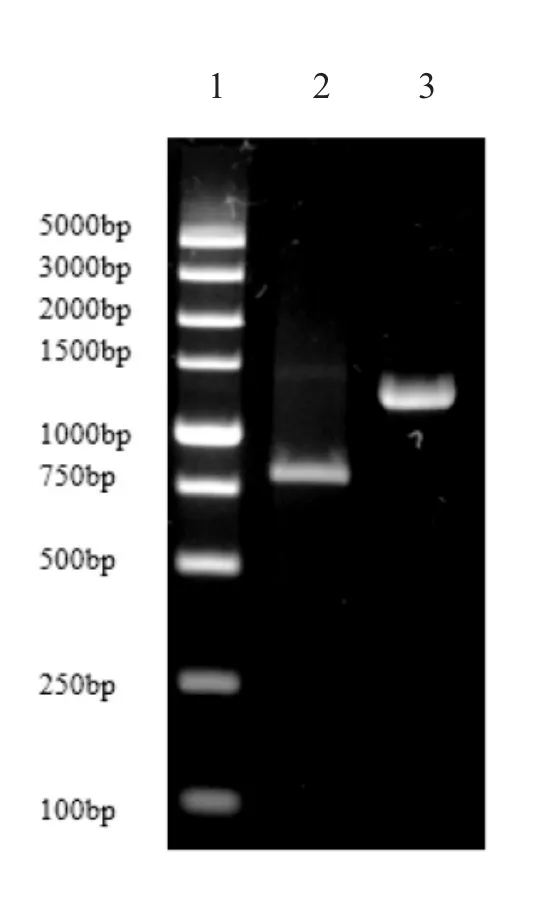
图1 SHVV P、N 基因的PCR 扩增产物Fig.1 The amplification products of P,N genes by PCR
2.2 重组表达质粒的构建及鉴定
将目的基因酶切后的片段纯化回收后,与酶切后的pGEX-5x-1 载体连接,构建融合GST 标签的原核表达重组质粒。经过测序和鉴定正确的重组质粒分别命名为pGEX-5x-1-N,pGEX-5x-1-P 分别经Sal I/Not I、EcoR I/Not I 双酶切后,利用1%的琼脂糖凝胶电泳对重组质粒进行鉴定(图2)。

图2 重组质粒的酶切鉴定Fig.2 Identification of recombinant plasmid by restriction enzyme digestion
2.3 重组蛋白的原核表达
将重组质粒pGEX-5x-1-N、pGEX-5x-1-P 分别转化至大肠杆菌感受态BL21(DE3)后,加入浓度1.0 mmol/L 的IPTG 诱导表达6 h,收集菌液及蛋白样品。利用SDS-PAGE 进行蛋白电泳检测,经过IPTG 诱导的和未经过诱导的空白载体pGEX-5x-1作为对照(图3),重组N 蛋白大小约为73 kD,重组P 蛋白大小约为57 kD,与预期相符。
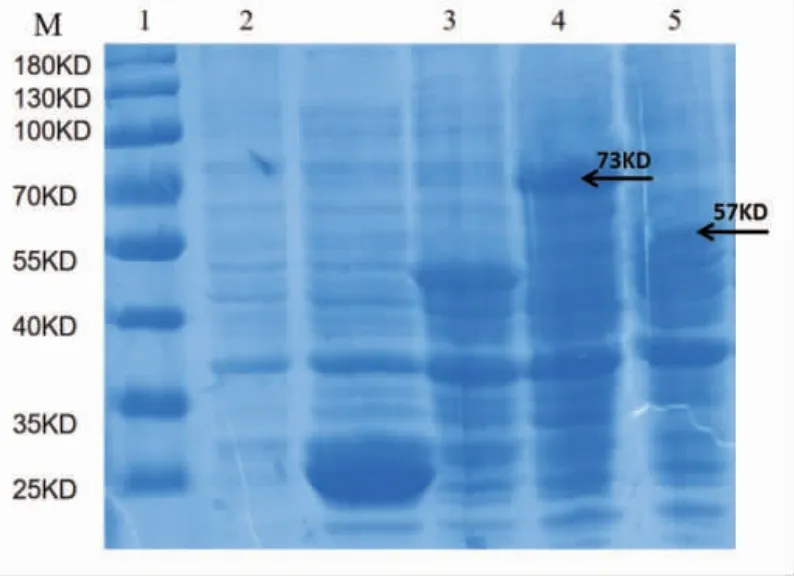
图3 重组质粒诱导表达蛋白的SDS-PAGE 检测Fig.3 Identification of recombinant plasmids prokaryotic expression by SDS-PAGE
通过蛋白凝胶回收试剂盒回收后,N 蛋白浓度为700 μg/mL,免疫所需总蛋白量为1 500 μg;P 蛋白浓度为300 μg/mL,免疫所需总蛋白量为300 μg。分别用N 蛋白作为抗原免疫兔子,P 蛋白作为抗原免疫小鼠,用ELISA 方法测得血清中特异性抗体的效价。当样品吸光值/阴性吸光值>2.1 时即认为是阳性。不同稀释倍数下实验组OD 值都在1 以上,且对照组OD 值很低,因此这两个多克隆抗体的效价均不低于1∶25 600(图4)。

图4 多克隆抗体效价的测定Fig.4 The titer of the anti-N,anti-P polyclonal antibody detected by ELISA
2.4 多克隆抗体的制备及Western-blot 检测
用0.1 MOI SHVV 感染SSN-1 细胞24 h 后收集细胞样品,对制备的多克隆抗体进行Western blot分析。N 蛋白与P 蛋白的多克隆抗体能够特异性地识别出SHVV N、P 蛋白的大小分别约为47 kD 和31 kD,且特异性良好,而对照组细胞没有检测到条带(图5)。
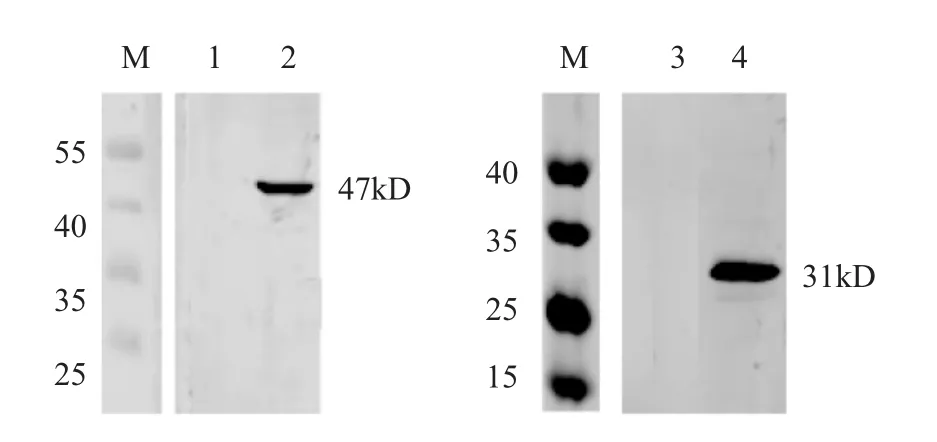
图5 N、P 蛋白多克隆抗体的Western blotting 分析Fig.5 Western Blot Assay of PcAbs against SHVV N protein and P protein
2.5 间接免疫荧光检测
将SSN-1 细胞接种在24 孔板中,长满单层后感染0.1 MOI SHVV16 h 后,用制备的N、P 蛋白的多克隆抗体为一抗,FITC 标记的商品化抗体为二抗,进行间接免疫荧光检测。结果:SHVV 病毒感染的SSN-1 细胞出现了明显的绿色荧光信号,而未感染组的SSN-1 细胞则没有相应的荧光信号,说明N、P 蛋白的多克隆抗体可以与SHVV 进行特异性结合,且N、P 蛋白主要分布在细胞质内(图6)。
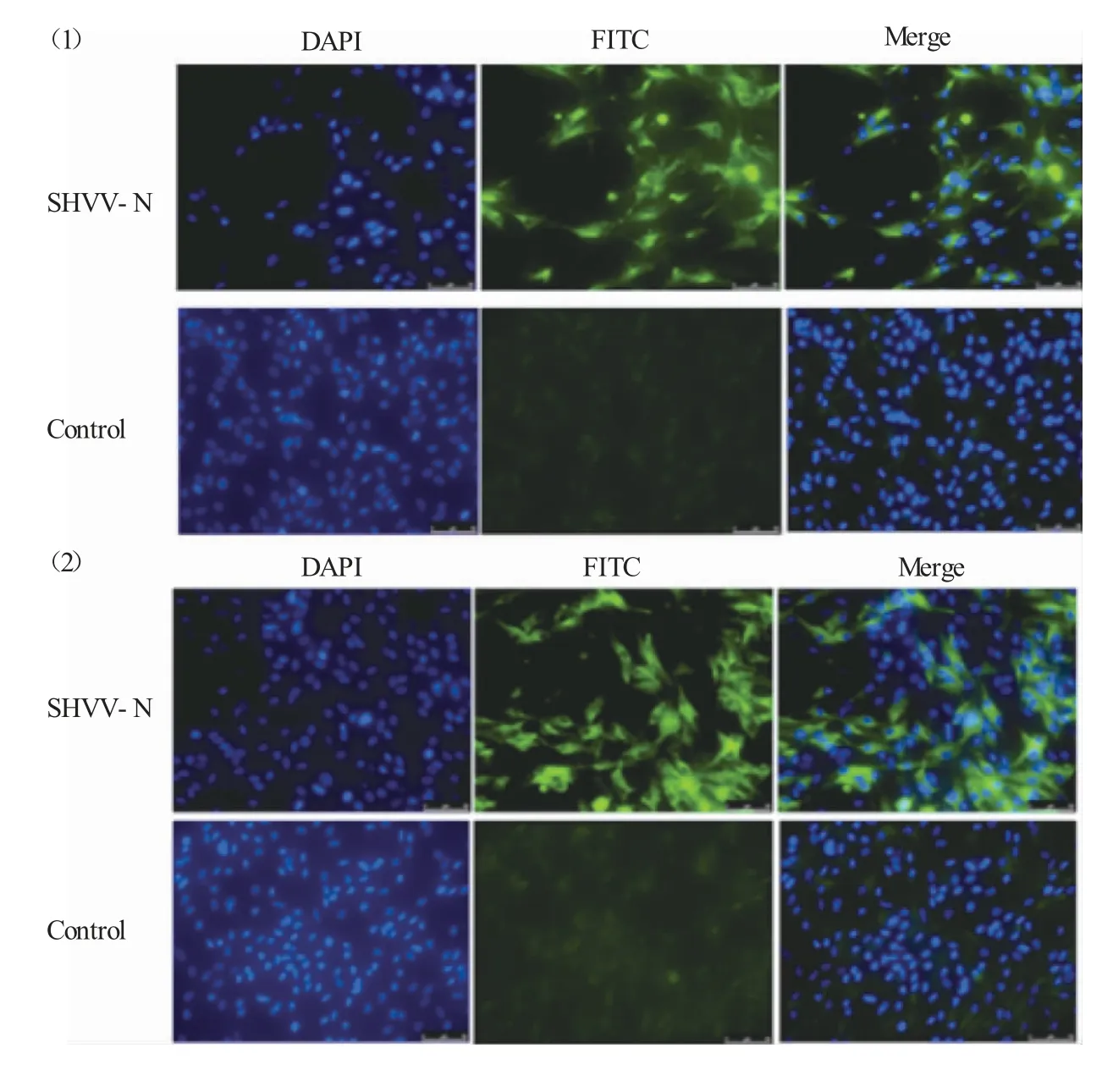
图6 SHVV 的N 蛋白(1)和P 蛋白(2)在细胞内分布Fig.6 Intracellular distribution of SHVV N protein(1)and P protein(2)
3 讨论
乌鳢水泡病毒SHVV 宿主广泛,除了能感染鳢科鱼类外还能感染鲈形目科的鳜(Siniperca chuatsi)等。鱼类感染SHVV 后,体表发黑,鳃苍白,胃肠道内没有食物,解剖观察有腹水[3,12],且感染病毒的鱼死亡率极高,目前为止缺乏对该病毒的诊断与有效防控方法。
弹状病毒科的病毒基因组RNA 主要编码N、P、M、G、L 5 种结构蛋白。核蛋白N 是含量最丰富、结构最保守的病毒结构蛋白[5],磷酸化蛋白P 是促使基因组从转录状态转换到复制状态的关键因子[13]。本实验扩增了N、P 蛋白的部分序列,将扩增产物克隆至原核表达载体pGEX-5x-1 中。pGEX 系列的原核表达载体带有GST 标签,而GST 标签能够促进目的蛋白正确折叠,有效提高蛋白表达可溶性[14]。因此分别构建融合GST 标签的重组质粒pGEX-5x-1-N 以及pGEX-5x-1-P,并转化感受态细胞BL21(DE3)。
大肠杆菌为原核生物,传代时间短,营养要求简单,遗传背景清晰,能够高效、迅速地表达外源蛋白等[15],但同时重组蛋白的原核表达常常形成包涵体,主要是在重组蛋白的表达过程中缺乏某些蛋白质折叠的辅助因子,或环境不适无法形成正确的次级键等原因导致。相较于可溶性蛋白,包涵体表达有以下有利因素:首先,可溶性蛋白在细胞内容易受到蛋白酶的攻击,包涵体表达可避免蛋白酶对外源蛋白的降解;其次包涵体表达降低了胞内外源蛋白的浓度有利于提高表达量。尽管包涵体的形成是目的蛋白表达量较高的表现[16],但大量包涵体的存在则对后续纯化造成不便。有研究将切胶纯化、过柱纯化、包涵体复性纯化等进行对比[17-19],结果证明包涵体复性纯化法适用于纯化低表达量蛋白,而相比于过柱纯化,切胶纯化的包涵体蛋白具有良好的抗原结合性,能够获得条带单一的目的蛋白,纯化效果较好。本实验选择直接切胶纯化。后续Western blotting 及间接免疫荧光检测分析结果表明,N 蛋白与P 蛋白的多克隆抗体可以与SHVV 特异性结合,特异性良好且N,P 蛋白主要分布在细胞质内。
多克隆抗体成本低、周期短、制备过程简单、可以识别更多的抗原、提高检测的灵敏度[20]。目前,已有很多鱼类病毒蛋白通过原核表达并成功构建多克隆抗体[21-24],均为后续研究提供基础资料。本研究制备了针对SHVV-N 及SHVV-P 的多克隆抗体,为建立该病毒免疫学检测方法奠定了基础,也期待后续更深入的研究。
猜你喜欢
环球时报(2022-09-20)2022-09-20
中国动物保健(2022年2期)2022-05-05
江西农业学报(2021年4期)2021-04-20
今日农业(2020年24期)2020-12-15
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
安徽医科大学学报(2015年9期)2015-12-16
小资CHIC!ELEGANCE(2015年15期)2015-09-01
西南医科大学学报(2015年1期)2015-08-22
中国当代医药(2015年9期)2015-03-01
现代检验医学杂志(2015年4期)2015-02-06
