
单原子电催化析氢催化剂的研究进展
2022-12-04 15:16:26王步祥舒庆
有色金属科学与工程 2022年5期
王步祥, 舒庆
(江西理工大学材料冶金化学学部,江西 赣州 341000)
经济发展致使能源需求剧增,伴随着煤炭等不可再生能源的过度使用, 能源和环境问题日益严峻,可再生能源的研发备受关注。 在能源需求方面,氢能因具有能量密度高、零排放和可循环利用等特点,未来大概率会替代传统化石燃料。 当前,制氢方式主要有4 种:化石燃料制氢、工业副产物制氢、电解水制氢(Hydrogen Evolution Reaction, HER)、生物质制氢。HER 是公认的最可持续、 最环保的从水中直接产生高纯氢的方法。
对于适用于HER 的催化剂而言,Pt 基催化剂一直是最佳选择[1-2]。 然而,成本高和稀缺性限制了Pt 基催化剂在HER 中大规模应用[3-4]。因此,人们一直致力于开发原料丰富、具有成本效益的高效HER催化剂[5-6]。 此外,对于以金属为主要活性组分的传统固体催化剂而言,只有极少数金属活性组分在催化反应过程中起催化作用,金属的利用效率远低于理想水平。 特别是对于贵金属,大剂量使用无疑增加了催化剂成本,不利于在工业化生产中应用。 单原子金属催化剂 (Single Atom Catalysts, SACs)的原子利用率在理论上高达100%, 是一种可高效提高原子利用率和反应活性的催化剂,近来受到了广泛关注。 2011 年,QIAO 等首次报道FeOx负载的单原子铂(Pt)催化剂可用于CO 的氧化反应过程,并在此基础上提出了单原子催化(Single Atom Catalysis,SAC)的概念[7]。
SAC 与纳米催化和亚纳米催化存在着明显的差异,这是因为当粒子分散度达到单原子尺寸时会出现诸多新的特性,如急剧增大的表面自由能、量子尺寸效应、 不饱和配位环境和金属—载体的相互作用等[8]。正是这些显著不同的特性赋予SACs 优越的性能:原子利用率高、100%的原子分散性、 均匀的反应中心、优异的选择性和高稳定性等[9-10]。然而,SACs 也存在不足, 这是由于金属粒子减小到单原子水平时,比表面积急剧增大会导致金属表面自由能急剧增加,从而在其制备过程和处于反应状态时极易发生团聚耦合形成大的团簇,导致催化剂失活[11]。因此,稳定性和活性是SACs 所面临的巨大挑战。
近年来,研究者尝试使用不同的方法制备活性与稳定性俱佳的SACs, 并采用多种先进的表征分析方法,以及结合理论计算推动SAC 的研究进程[12-14]。本文首先列举了SACs 的常规制备方法, 如热解法(高温热解、热解迁移)、沉积法(化学气相沉积法、原子层沉积法、电化学沉积)、液相合成和刻蚀法,并比较了上述制备方法的优缺点;列举了常见的SACs 的表征分析方法, 以及SACs 在HER 领域的研究进展情况;最后,指出目前SACs 在应用于HER 时面临的挑战,并提出了一些可能的改进方法,以期为设计廉价高效的用于HER 的SACs 提供有价值的参考。 常用的单原子催化剂制备、表征及密度泛函计算方法如图1 所示。
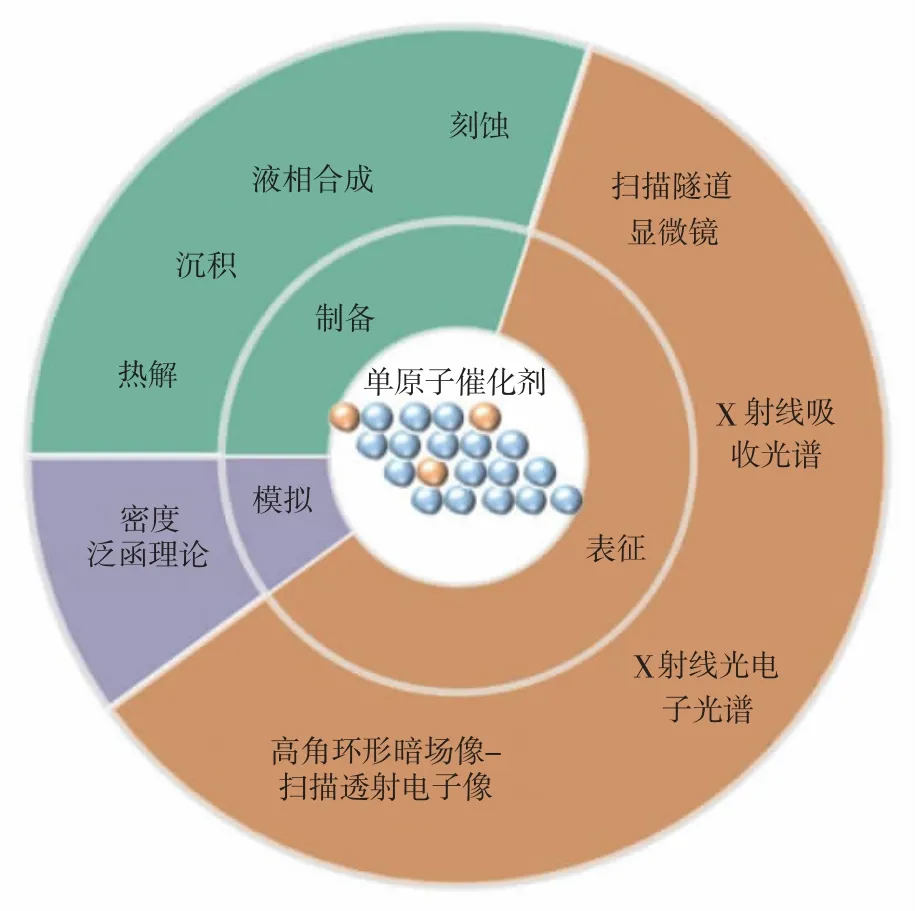
图1 常用的单原子催化剂制备、表征及密度泛函计算方法Fig. 1 Common preparation, characterization and density functional calculation methods of single-atom catalyst
1 合成方法
目前, SACs 通常通过热解、沉积、液相合成和刻蚀等方法制备而成。
1.1 热解法
热解法是一种简便易行且广泛使用的SACs制备方法, 但在热解过程中无法避免高能耗的问题。 通常是将金属离子浸渍到载体上,然后在高温惰性气体氛围下热解成SACs[15-16]。 其中,氧化石墨烯(Graphene Oxide, GO) 和 碳 纳 米 管(Carbon Nanotubes, CNTs) 由于含有大量可捕获和牵引金属阳离子的含氧官能团, 因而成为热解法制备SACs 时广泛使用的2 种碳基材料。首先将贵金属或过渡金属原子分散到氮掺杂的GO 或CNTs 中,然后在高温退火条件下将混合物碳化,形成稳定的SACs[17]。LI 等报道了单原子Ni(SA Ni)促进Pt 电催化的最新研究成果,通过合成的平均长度约为300 nm、 直径约为4.5 nm 的富镍PtNi 合金纳米线,在70 mV 过电位(η),pH=14的反应条件下, 质量活性可达到11.8 A/mg Pt[18]。ZHANG 等报道了一种将单原子Pt 从前驱体碳球表面迁移至其内部的方法, 实现了孤立Pt 原子在介孔碳基质(Mesoporous Carbon Matrix, PCM)中的牵引和稳定, 合成了活性中心高度分散的Pt@PCM 电催化剂。 以0.5 mol/L H2SO4溶液为电解液,利用三电极体系对Pt@PCM 的HER 性能进行了测试。 结果发现:Pt@PCM 对HER 具有很高的催化活性,其起始电位(η。)接近HER 的热力学电位(即0 Vvs. RHE),与商用20% Pt/C 的η。 相当。 通过计时电流i-t曲线可知,Pt@PCM 在酸性介质中5 h 后的电流衰减小于起始电流的5%,表现出优异的稳定性[19]。其次,金属有机骨架化合物 (Metal Organic Frameworks, MOFs)作为一类新型多孔材料[20-22], 也可以作为单原子的载体,通过热解法制备SACs。 如CHEN 等通过将钨前驱体(WCl5)封装在MOF 骨架(UiO-66-NH2)中,然后进行热解, 合成了钨单原子电催化剂 (WSAC)。 在0.1 mol/L KOH 溶液中进行的HER 性能测试表明:W-SAC 与商用Pt/C 几乎具有相等的活性。 此外,研究人员对W-SAC 进行了耐久性测试,测试结果显示,W-SAC 经10 000 次CV 循环测试,活性并没有明显降低,这表明其具有用于实际HER 的潜力[23]。 MOFs 作为SACs 的载体有以下优点:不同种类的金属离子可以通过不同的有机前体进行桥联; 掺杂的杂原子也可以通过热解固定在SACs 上[24]。
通过热解过程中的原子迁移作用, 同样可实现SACs 的制备[25-26]。 但此类方法不仅需要可移动的金属物种,而且需要一种能够捕获可移动金属物种的支撑材料[27]。 通常是在惰性气氛和高于900°C 的环境中进行,首先将贵金属纳米颗粒转变为热稳定的金属单原子;随后,辅助气体将金属单原子运送至多孔载体上[28]。QU 等以有缺陷的石墨烯 (Defective Graphene, DG)锚定铂单原子(Pt SAs)的方式制备了Pt SAs/DG 催化剂, 通过高温热解二氰胺产生的氨气促进Pt 网上的Pt 转变为Pt 原子,Pt 原子被具有丰富缺陷的DG捕捉,并在热迁移的作用下陷入DG 表面的双缺陷位点中, 形成Pt-C4的配位结构, 实现从块体Pt 到Pt SACs 的一步转化。 该催化剂表现出超越商用Pt/C 的HER 性能。 电流密度为10 mA/cm2时,Pt SAs/DG 的η为23 mV,低于商用Pt/C(38 mV)[29]。 热解原子迁移俘获法可实现非贵金属(铜、镍和钴)一步转化为SACs。
1.2 沉积法
通过化学气相沉积法 (Chemical Vapor Deposition, CVD) 将金属原子沉积到选定的支撑衬底上,是常用的SACs 合成方法[30]。 利用气态的先驱反应物,通过原子、分子间化学反应,使得气态前驱体中的某些成分分解,并在基体上形成薄膜。QIU 等报道了通过CVD 法合成用于HER 的单原子镍掺杂纳米多孔石墨烯催化剂,该催化剂的塔菲尔(Tafel)斜率约为45 mV/dec, 说明镍的掺杂极大地改变了石墨烯的化学活性。在运行120 h 后,催化活性保持约90%,证实了单原子镍掺杂纳米多孔石墨烯催化剂具有高稳定性[31]。 HAN 等提出了一种利用一步CVD 法在1T-WS2单分子层上实现高度分散的单原子钒催化剂(V SACs@1T-WS2)制备的方法。 以0.5 mol/L H2SO4溶液为电解液,通过三电极装置进行了HER 性能测试, 线性伏安扫描表明其在10 mA/cm2的电流密度下,η 为185 mV, 而钒的负载仅为1.8~6.5 μg/cm2。 在100 mV 时,塔菲尔斜率为61 mV/dec,对应的周转频率(Turnover Frequency,TOF)值为3.01 s-1,显示出良好的催化性能[32]。 尽管CVD 法是制备杂原子掺杂石墨烯的有效方法, 但低产量的石墨烯极大地限制了其实际应用。 此外,无论是预沉积的金属膜,还是预抛光的金属箔,都可能导致金属残留,从而污染SACs。
原子层沉积法(Atomic Layer Deposition, ALD)是一种通过将气相前驱体脉冲交替地通入反应器并在沉积基体上通过化学反应吸附而形成沉积膜的方法[33-35]。 FANG 等利用ALD 法将Pt 原子孤立地分散在掺氮的碳(Nitrogen-Carbon, N-C)载体中,形成高度均一的Pt1/N-C, 电化学测试证实其具有优异的HER 性能。 在1.0 mol/L KOH 和0.5 mol/L H2SO4溶液中,Pt1/N-C 的η 均低于商用Pt/C 催化剂, 分别达到了极低的46 mV 和19 mV,TOF 更是超过Pt/C一个数量级。 并且,Pt1/N-C 还表现出极佳的稳定性,在20 h 稳定性测试后,其活性几乎没有衰减[36]。ZHANG 等通过ALD 法将Pt 单原子负载至氮掺杂CNTs 上,随着载体中的类吡咯氮位点增加,Pt 催化剂的HER 活性随之提高。与商用Pt/C 催化剂相比,具有更高类吡咯类氮含量的CNTs 负载Pt 单原子形成的催化剂表现出更高的HER 性能, 其质量活性比商用Pt/C 高47 倍[37]。 ALD 可以精确地控制沉积参数,使得金属原子在载体表面均匀沉积且重复性好,而且ALD 亦可精确控制不同材料以原子层的方式生长,形成具有不同形貌的复合物。然而,ALD技术面临着2 个主要挑战,即低金属负载量和金属位点的不均匀性。 同时,ALD 还受到产量低、设备成本高和前驱体的限制, 这些都不利于ALD 的大规模应用[38-40]。
电化学沉积法是在一定电压下, 原子从阳极溶解,再沉积到阴极上的方法。 该方法具有操作简单、条件温和、成本低和精确可控等特点,因此被广泛应用于纳米材料的制备和电镀等工业生产中[41-44]。ZHANG 等报道了一种利用电化学沉积制备SACs 的普适性方法。 阴极沉积的Ir1Co0.8Fe0.2Se2/C 在1 mo l/L KOH 中进行HER 测试, 仅需8 mV 的过电位即可达到10 mA/cm2的电流密度,远低于商用Pt/C[45]。ZHANG 等通过电化学的方法将Pt 单原子固定在由CoP 的纳米管(Nanotube, NT)阵列支撑泡沫Ni(Nifoam, NF)形成的载体上,合成了Pt-NT-NF 催化剂。 在中性介质中进行HER 测试,10 mA/cm2的电流密度下, 表现出24 mV 的超低η,塔菲尔斜率仅为30 mV/dec。 在η=50 mV 时,Pt 的质量活性为70 A/mg,大约是商用Pt/C的4 倍,其电催化稳定性也优于商用Pt/C;由计时电流曲线可知,其高活性保持了24 h[46]。 然而,这种常规的电沉积工艺不可避免地会形成覆盖率不均匀的多层体相结构。 为了克服这一困难,有研究者采用表面限制的欠电势沉积技术 (Underpotential Deposition,UPD) 制备单层金属结构[47]。 如SHI 等制备得到Pt SAs/MoS2催化剂,在酸性介质中进行HER 测试,相比于纯MoS2基底,Pt SAs/MoS2表现出优异的HER 电催化活性,10 mA/cm2的电流密度对应的η 为59 mV,质量活性是商用Pt/C 的114 倍。 在η 为100 mV 条件下,Pt SAs/MoS2的塔菲尔斜率低至31 mV/dec,TOF 值高达47.3 s-1,高于绝大多数的SACs。 此外,该催化剂电催化稳定性良好[48]。 该方法可在常温常压下实现可控、快速地合成原子分散的金属催化剂, 并且具有一定的普适性[49-51]。 然而,这种电沉积工艺也有缺点,如电镀不均匀。
1.3 液相合成法
SACs 也可以在液相中通过各种相互作用合成,如共价键、p-p 堆积、静电吸引或螯合反应等[52-54]。CHENG 等报道了Pt/TiO2-OVs 催化剂,在η 为50 mV时,该催化剂具有较优的质量活性(62.34 A/mg)和最高TOF (56.1 s-1), 比商用Pt/C 高18.7 倍和5.56 倍[55]。WANG 等首先采用限域法合成了单层WO3·H2O(ML-WO3·H2O)纳米片,然后将Pt 单原子(Pt-SA)固定在单层WO3(ML-WO3)纳米片,成功构建了Pt-SA/ML-WO3。实验结果显示,Pt-SA/ML-WO3具有优异的电催化性能,仅需22 mV 的过电位即可实现10 mA/cm2的电流密度,塔菲尔斜率低至27 mV/dec,同时在η 为50 mV 时,具有87 s-1的超高TOF,以及高稳定性。 特别值得注意的是,Pt-SA/ML-WO3在η 为50 mV 条件下具有87 A/mg 的超高质量活性,是最先进的商用Pt/C(0.54 A/mg)的160 倍[56]。 液相法适用大多数载体,且操作简单, 这为相应SACs 的大规模生产提供了机会[57-60]。 但是,包含在前体材料中的孤立金属原子因高表面能而具有强烈的聚集趋势,所以这种方法并不能制备高负载量的SACs[61]。
1.4 刻蚀法
组装/刻蚀法是一种提高电催化剂析氢性能的常用方法, 通过刻蚀可改变材料的形貌及结构,增大材料的比表面积并暴露更多的活性位点,随后负载活性物质,从而制备高负载量的SACs[62-64]。 QI 等成功制备了一种新型界面催化剂, 这一催化剂由MoS2纳米片与金属单原子钴(SA Co)通过共价结合组成(SA Co-D 1T MoS2)。 组装过程中,由于Co 与MoS2之间的晶格失配和Co-S 之间形成强共价键,作为单原子催化剂载体的MoS2会发生由2H 向扭曲的1T 相的转变,该现象进一步阐释了Co 单原子与载体之间的相互作用关系。 在酸性电解液中,其具有类商用Pt 的HER 活性,且具有优异的稳定性。HER 测试中, 钴负载量为3.54%的SA Co-D 1T MoS2表现出极小η。(42 mV),远小于其他非贵金属催化剂,甚至与Pt/C(Pt 负载量10%)的η。 相当[65]。尽管组装/刻蚀法制备的电催化剂具有负载量高和性能好的优点,但仍有许多问题。 首先,由于刻蚀会暴露了大量的金属原子, 催化剂处于热力学亚稳态,容易氧化,导致其失去活性。 其次,目前主流的刻蚀溶液仍具有高毒性和污染性。
用于HER 的SACs 的性能比较分析如表1所列。
从表1 可知:通过不同方法制备而成的SACs 具有不同的η。

表1 单原子催化剂的性能比较分析Table 1 Comparison and analysis of the performance of single atom catalysts
电解水反应包括阴极还原反应(即HER)和阳极氧化反应(即析氧反应(Oxygen Evolution Reaction,OER))。 OER 和HER 分别如式(1)和式(2)所示,总的水分解反应如式(3)所示。


因此,理论上水分解产生氢气的电位是0 V。 实际上, 不同制备方法会产生不同类型的单原子位点,这些单原子位点具有不同的配位构型和电子特征,最终影响SACs 的结构—催化关系,从而导致不同方法制备的SACs 具有不同的η。 因此,有必要在原子尺度上识别SACs 的活性位点, 了解SACs 的结构—催化关系。
2 表征分析和密度泛函计算
2.1 SACs 表征技术
为了探索SACs 的结构—催化关系和实现高性能催化剂的合理设计,在原子尺度上识别活性中心并监测其配位环境是非常必要的。许多先进的表征和分析方法已被用于深入研究SACs 的结构—催化关系,包括扫描隧道显微镜 (Scanning Tunneling Microscope,STM)、高角环形暗场像—扫描透射电子像(High-Angle Annular Dark-Field Imaging-Scanning Tunneling Microscope,HAADF-STEM)、X 射线光电子能谱(X-ray Photoelectron Spectroscopy,XPS)、X 射线吸收光谱(X-ray absorption spectroscopy,XAS)等[66-68]。
STM 可以直接揭示SACs 的形貌、 电子特征,避免了从光谱或化学测量中间接分析化学信息的主观性。 STM 以波长极短的电子束作为照明源,用电磁透镜聚焦成像。WANG 等通过STM 对单层ML-WO3纳米片进行了结构分析,STM 图像表明Pt-SA/ML-WO3具有不规则片状形态。 从ML-WO3到Pt-SA/MLWO3,没有出现严重的烧结现象[56]。 传统的SEM 或TEM(Transmission Electron Microscope, TEM)技术的分辨率有限,无法观察单原子的具体形态;具有球面像差校正的HAADF-STEM 可直观观测到单原子在载体上的分散情况,极大地推动了SACs 的研究进程。 QI 等通过HAADF-STEM 对原子Co 与扭曲的1T MoS2纳米片共价结合组成的界面催化剂SA Co-D 1T MoS2进行分析, HAADF-STEM 图像证实了结合界面处MoS2从2H 转变为D-1T 相, 且SA Co-D 1T MoS2和2H MoS2之间存在明显界面,Co 是位于Mo 原子的上方,而不是替代Mo[65]。TIAO 等成功合成了N 掺杂空心多孔碳 (Nitrogen-Doped Hollow Porous Carbon, HC)负载的Co 单原子(SA Co/HC),通过HAADF-STEM 图像可以观察到高密度的亮点,显示了原子级Co 位点的分散情况[69]。 与SEM 或TEM 对比,HAADF-STEM 能获得更清晰的SACs 形貌和组分信息,但其电子束很难约束,球差校正难度更大。
XPS 技术可提供SACs 表面的化学信息, 如元素组成、元素含量及其价态。 XPS 技术通过用X 射线(1 000~1 500 eV)照射样品,SACs 的价电子或内壳层电子可以被激发。 同时,测量从SACs 表面逃逸的电子的数量和动能,测得的发射电子的数量和动能分别反映了表面元素的含量和化学状态信息。ZHANG 等通过XPS 对Pt@PCM 的结构进行了分析,PCM 的N1s 光谱证明基质中存在石墨、 吡啶和吡咯类氮物种,与上述物种对应的特征峰分别在结合能为401.6、400.6、398.6 eV 处出现。 吡啶中存在的N 可以作为Pt 原子的固定点,Pt 原子的改性增加了N1s 的平均结合能,同时,吡啶中N 的比例也得到了提高。上述结果证明了孤立的Pt 原子和碳基体之间存在相互作用,以及孤立的Pt 中心和配位的N 原子之间的电子转移。 此外,在Pt@PCM 中Pt 4f的XPS 谱中可以观察到2 个峰,分别出现在结合能为71.28 eV 和74.53 eV 处,这表明在Pt 原子和吡啶中C/N 之间形成了配位键, 而在70.89 eV 和74.23 eV 处不存在与Pt-Pt 键相对应的峰, 证实没有形成Pt 团簇粒子[19]。XPS 分析所得到的信号只能确定元素有无以及大致位置,并且X 射线的穿透深度有限,得不到更准确的元素化学态和分子结构信息等。
与XPS 技术对比,XAS 技术具有亚原子分辨率和超高电子结构敏感度,其X 射线(0.1~100 keV)能将特定原子轨道上的电子激发到自由态或未占据的能级, 因而XAS 技术能更准确表征SACs 的化学状态和几何结构。XAS 又可分为2 种模式:扩展的X 射线吸收精细结构 (Extended X-ray Absorption Fine Structure, EXAFS)和X 射线吸收近边缘结构(X-ray Absorption Near Edge Structure, XANES)。 SACs 的局部结构信息可以由EXAFS 确定, 例如原子间距、配位数、邻近原子的数量和类型等。XANES 数据分析可提供中心金属原子的化学状态和几何信息,例如确定价态、表征d-带特性、测定配位电荷、提供包括轨道杂化、配位数和对称性等。 通常选定SACs 中需要测试的某种元素, 用特定的X 射线光子能量扫描,将内壳电子激发到未占据的能级, 从而导致某些能量(吸收边)急剧上升。 每个吸收边与SACs 中存在的特定原子有关,并因为周边原子的能量散射形成一系列振荡结构。 以X 射线光子能量作为变量,测定SACs的X 射线吸收系数。 其中,硬X 射线(能量较高的X射线,波长在0.01~0.1 nm 之间)具有研究原位电化学反应动态过程的能力,有利于建立更精确的结构—催化关系。 FANG 等利用ALD 在金属有机骨架(MOF) 衍生的N-C 骨架上合成了原子分散的Pt 催化剂,将其应用于HER 过程时,通过XAFS 技术在原子水平上确定了单个原子向近自由状态的演化过程。通过XANES 光谱中的白线峰的吸收边能量和面积积分,计算定量确定铂的价态和d 带空穴数,从而获得电子结构演化的定量信息。 测试显示,在+0.5~+0.15 V和-0.07 V 的电势下,Pt 的平均价态分别从+1.89 降低到+1.17 和+1.12,这意味着Pt 的价态向金属态降低,直到接近零价, 验证了在工作状态下Pt 和基底之间存在弱相互作用。 EXAFS 进一步确定了单原子Pt 配位构型的演化。 在相应的外加电位下,Pt1/N-C 的EXAFS 谱都在约0.16 nm 处呈现一个单一的主峰,这可归因于Pt 原子与C/N/O 原子之间的配位。然而,当施加的电位分别从+0.5 V 负移至+0.15 V 和-0.07 V时,傅里叶变换峰的强度衰减了13%和35%,这表明在工作条件下Pt 位点的局部几何结构有明显的变化。 振荡峰的强度随着电势的增加而降低,证实了Pt 原子周围的动态无序结构。 值得强调的是,EXAFS 和XANES 分析是从相同的SACs 中获得的, 对配位环境和几何/电子结构有不同的表现形式, 但也由此检测出SACs 的结构和化学信息应一致,可以相互验证[36]。XAS 技术有助于理解SACs 的结构—催化关系,但XAS 技术的实验装置和条件很难控制,特别是在软X 射线(能量较低的X 射线,波长在0.1~10 nm 之间)范围内,这仍然是一个很大的挑战。
2.2 SACs 电子特性的密度泛函计算
密度泛函理论(Density Functional Theory, DFT)作为处理多粒子体系的近似方法在物理、材料、化学和环境等领域广泛应用[70]。 DFT 通过多种近似方法,将难以解决的包含电子—电子相互作用的问题简化成无相互作用的问题, 将所有误差单独放进一项中,再对这个误差进行分析。 DFT 可从原子或者分子尺度理解并且调控物质的性质。 首先假设催化剂的结构,然后模拟不同表面原子间的相互作用,探究纳米表面的催化活性, 预测出催化剂的种类以及活性位点。 基于DFT 的科学计算模拟方法不仅可以研究现有SACs 的催化性能, 而且可以预测并设计新的SACs。 WANG 等为了进一步阐明Pt-SA/ML-WO3的催化活性, 利用DFT 计算了催化剂模型的氢吸附自由能(ΔGH*)。通常,接近热中性的ΔGH*值(ΔGH*≈0 V)对应于高活性, 计算结果显示WO3、WO3-VO/W、Pt/WO3-VW 和Pt/WO3-VO(VO 为氧空位数量)的ΔGH*值为0.55 eV、0.38 eV、-0.17 eV 和0.10 eV, 表明O/W空位的存在和Pt 原子的掺入都可以增强SACs 对氢的吸附能力。 特别是, Pt 原子取代W 原子使得Pt/WO3-VW 的ΔGH*非常接近理想金属Pt(111)(-0.09 eV),DFT 计算表明,Pt 和载体ML-WO3之间存在很强的相互作用,这极大地调整了催化剂的电子结构,赋予其很强的导电性和合适的ΔGH*值,并产生优异的电催化活性[56]。 LI 等通过使用负载在N 掺杂碳纳米片(N-doped Carbon Nanosheets, NCNS)上的Pt 单原子作为催化剂(Pt1/NCNS),利用原子层沉积(ALD)技术合成了Co SAC。 DFT 计算表明,Pt 原子作为催化剂调节Co 原子在ALD 中的吸附行为,促进Co (Cp)2的解离,CoCp 与吡咯N4位点结合形成Co 单原子,而不是纳米粒子。这种合成策略也可以扩展到合成FePt/NCNS 和NiPt/NCNS。 在电化学测试中,NiPt/NCNS 比Fe 和Co 的SACs 表现出更好的催化活性,这与DFT 预测一致[71]。 ULLAH 等使用密度泛函理论方法设计和研究了石墨炔(Graphyne, GY)表面负载过渡金属(Fe、Co、Ni、Cu 和Zn)的SACs。不同SACs 的ΔGH*表明,锚定在GY 载体上的Ni/GY 具有最高的热力学稳定性和最好的HER 催化活性[72]。 DFT 方法是通过将电子的密度分布函数作为描述体系所有性质的唯一变量来计算粒子体系的各种性质,具有计算量小的优点,但是也会受到简化处理的制约,必然会出现误差。 在近似的基础上修正也许能够接近实际的数值, 但是理论基础依然还是在同一个维度上。 DFT 适用范围相对有限,模拟效果仍需实验结果佐证。
通过多种表征方法和理论计算不仅有利于指导SACs 的设计, 更有助于揭示SACs 在复杂催化环境中的化学反应机制。
3 结束语
对用于HER 的SACs 的研究进展进行了综述,首先对SACs 的制备方法进行总结, 重点介绍SACs的热解、沉积、液相合成和刻蚀4 种制备策略;然后对SACs 的表征技术进行了总结,包括扫描隧道显微镜、高角环形暗场像—扫描透射电子像、X 射线光电子光谱、X 射线吸收光谱, 并介绍了密度泛函计算在解析SACs 电子特性方面的应用。
尽管SACs 在HER 的研究取得了一定的进展,但目前还停留在实验室小试阶段, 达不到工业化标准。 有关SACs 的下一步研究应集中在以下6 个方面: ①在确保材料稳定的基础上提高其电催化活性,发展最佳改良技术;②复合材料中考虑引入二维材料为助催化剂, 降低单原子巨大的表面能和迁移能力,防止在制备过程和处于反应状态时发生团聚耦合形成趋于稳定的纳米颗粒,导致催化剂失活;③开发新的、更高效的支撑材料,提升基底材料与金属之间的相互作用以及稳定性,从而促进催化性能;④利用过渡金属和非贵金属制备高效、稳定、低成本的SACs;⑤利用新的表征手段和理论模拟运算技术,从微观和瞬态两个方面阐明电催化机理,特别是结合实验结果和密度泛函计算在分子尺度上探究HER 机理;⑥明确单原子电催化剂的结构和电解水制氢活性之间的构效关系,为高效电催化剂和电催化体系的设计提供必要的理论指导。
猜你喜欢
睿士(2023年10期)2023-11-06 14:12:16
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 06:42:32
小学科学(学生版)(2021年3期)2021-04-13 08:26:18
小哥白尼(趣味科学)(2020年9期)2021-01-18 06:12:42
沉积与特提斯地质(2019年1期)2019-07-16 08:41:30
沉积与特提斯地质(2018年4期)2018-07-16 08:27:30
南方文学(2016年4期)2016-06-12 19:58:50
Coco薇(2015年5期)2016-03-29 23:14:09
