
高效液相色谱-串联质谱法测定猪肉中可的松和氢化可的松残留量的不确定度评价
2022-12-03 09:24:12程用斌王舒婷吴晓红车彦卓
中国兽药杂志 2022年11期
张 婷,程用斌,王舒婷,吴晓红,凌 莉,李 嘉,李 祯,车彦卓,柏 旭
(陕西省农业检验检测中心,西安 710000)
可的松和氢化可的松属于糖皮质激素类[1],是可随动物体应激反应而提升分泌水平的内源性激素,不需要制定最大残留量[2-4],人工合成的可的松和氢化可的松属于世界反兴奋剂中心禁止运动员使用的药物,规定其在动物组织中的残留限量均不得超过30 μg/kg[5]。
为了更合理、更科学地表示测量结果,实验室需根据相关认可的技术规范要求进行不确定度评价[6],本文基于中心自建方法《动物源性食品中克伦特罗等51种兴奋剂的兴奋剂检测方法 液相色谱-串联质谱法》和CNAS-GL106《化学分析中不确定度的评估指南》[7],在实践过程发现51种兴奋剂中可的松和氢化可的松的检出率最高,故筛选出这两项具有代表性的食源性兴奋剂进行研究,开展平行测试进行不确定度分析,为检验结果的准确性及可靠性提供技术依据,同时为本实验室自建方法的建立提供数据支撑,为其他实验室采用内标法评定不确定度提供参考。
1 材料与方法
1.1 仪器与设备 TQ-S高效液相色谱-质谱仪(美国Waters仪器公司);PL402-L型电子天平(瑞士METTLER TOLEDO公司);DU2A12FV超纯水仪(泽布拉公司);KQ-超声波清洗器(昆山市超声仪器有限公司);AutoEVA-20Plus全自动氮吹浓缩仪(睿科仪器公司)。
1.2 试剂与材料 可的松标准物质(不确定度:2%,k=2;纯度98.5%)(北京曼哈格生物科技有限公司);氢化可的松标准物质(不确定度:3%,k=2;介质:甲醇;浓度99.8ug/ml)(北京曼哈格生物科技有限公司);氢化可的松-D3标准物质(不确定度:5%,k=2;介质:乙腈;浓度100 ug/mL)(阿尔塔科技有限公司);甲醇(质谱纯,美国Thermo Fisher);乙腈(色谱纯,美国Thermo Fisher);甲酸(美国Thermo Fisher);无水硫酸钠(分析纯)。
除另有说明,试验中所用制剂均按GB/T 603的规定制备。水为符合GB/T 6682规定的一级水。
1.3 实验条件
1.3.1 前处理方法 称取试样5.00 g(精确至±0.01 g)于50 mL具塞离心管中,加入100 μL混合同位素内标中间液,涡旋混匀,加入10 mL1%甲酸乙腈,涡旋振荡10 min,加入3 g无水硫酸钠,涡旋振荡2 min,8000 r/min离心5 min,取上清液于另一干净的50 mL具塞离心管中,剩余部分再用10 mL1%甲酸乙腈重复提取一次,合并上清液,精密移取2 mL上清液于10 mL离心管中,于40 ℃下氮气吹干,准确加入1 mL乙腈-水(50+50,v/v)溶液溶解残渣,涡旋振荡1 min,过0.22 μm尼龙滤膜,供液相色谱-串联质谱仪测定。
1.3.2 色谱条件 色谱柱:Waters BEH C18(2.1×100 mm,1.7 μm);流动相条件:A相:0.1%甲酸水,B相:0.1%甲酸甲醇;梯度洗脱:0~1 min,维持95%A;1~2 min,A相线性变化至80%;2~6 min,A相线性变化至40%;6~8 min,A相线性变化至20%;8~8.5 min,A相线性变化至10%;8.5~9.7 min,A相维持10%A;9.7~9.75 min,A相线性变化至0%;9.75~12.75 min,A相维持0%;12.75~12.8 min,A相线性变化至95%;12.8~15 min,A相维持95%。流速:0.35 mL/min,柱温:40 ℃。
1.3.3 质谱条件 离子化模式:电喷雾电离(ESI);正离子模式;电离电压:3200V;离子源温度:150 ℃;扫描模式:多反应监测(MRM);干燥气温度:550 ℃;碰撞气:氩气。质谱参数见表1。

表1 质谱扫描参数
1.4 标准溶液配制 称取可的松对照品约10 mg,精密称定。用甲醇溶解并定容至10 mL,配成浓度为1000.8 μg/mL标准储备液,然后精密量取20 μL可的松标准储备液、400 μL氢化可的松标准溶液至10 mL容量瓶,用甲醇分别配制成2.0、4.0 μg/mL的混合标准中间液。精密量取1 mL混合标准中间液至10 mL容量瓶中,用甲醇定容,配制成200 ng/mL可的松、400 ng/mL氢化可的松混合标准工作液。同时精密量取1 mL氢化可的松-D3内标溶液至100 mL容量瓶中,用甲醇配制成1 μg/mL的内标标准工作液。标准溶液均置于-20 ℃保存。
1.5 标准曲线制作 取6份空白样品,按照1.3.1前处理步骤得到空白残余物,分别加入10 mL乙腈-水(50+50,v/v)溶解制成空白基质溶液,分别精密量取0、100、200、300、400、800、1600 μL混合标准工作液于7个10 mL容量瓶中,各加入100 μL的内标标准工作液,用空白基质溶液定容至刻度,配成氢化可的松浓度分别为0、4、8、12、16、32、64 ng/mL;可的松浓度分别为0、2、4、6、8、16、32 ng/mL的混合标准系列溶液,过0.22 μm尼龙滤膜,供液相色谱-串联质谱仪测定。
2 结果与分析
2.1 不确定度数学模型建立
式中:X—试样中各待测物的含量,μg/kg;C—标准工作溶液待测组分的含量,ng/mL;Ci—测试液中内标物的浓度,ng/mL;A—测试液中待测组分的峰面积;Asi—标准工作液中内标物的峰面积;V—定容体积,mL;Csi—标准工作溶液中内标物的浓度,ng/mL;Ai—测试液中内标物的峰面积;As—标准工作液中待测组分的峰面积;m—样品的称样量,g;f—稀释倍数;R—方法回收率。
2.2 不确定度来源分析 根据检测方法以及已建立的数学模型来分析,猪肉中可的松、氢化可的松的含量不确定度分量的主要来源为:Urel(1)样品称量引入的不确定度;Urel(2)样品定容引入的不确定度;Urel(3)标准物质引入的不确定度;Urel(4)回收率引入的不确定度;Urel(5)方法重复性引入的不确定度。具体来源分析如图1。

图1 不确定度来源
2.3 不确定度分量的评定


2.3.3 标准物质引入的不确定度 (1)标准物质纯度引入的不确定度:根据标准物质证书,按扩展因子,k=2,标准物质纯度的相对标准不确定度为:U(3.1)=u/(p×2),见表2。

表2 标准物质纯度引入的不确定度
(2)标准品称量引入的相对不确定度

(3)标准溶液配制过程引入的不确定度
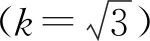
①标准储配液配制引入的相对标准不确定度

②标准中间液配制引入的相对标准不确定度

③标准使用液引入的相对标准不确定度

④加入内标引入的不确定度
采用SPSS 21.0软件对数据进行分析处理,计量资料以(均数±标准差)表示,采用t检验;计数资料以(n,%)表示,采用χ2检验,以P<0.05表示差异具有统计学意义。

⑤标准曲线配制引入的不确定度
配制标准工作曲线,吸取标准溶液时,使用A级0.1 mL分度吸量管8次,A级0.2 mL分度吸量管1次,A级1 mL分度吸量管3次,A级2 mL分度吸量管1次,A级10 mL容量瓶7次。标准曲线配制中体积引入的相对标准不确定度见表3。则此部分的合成相对标准不确定度为U(3.22)=
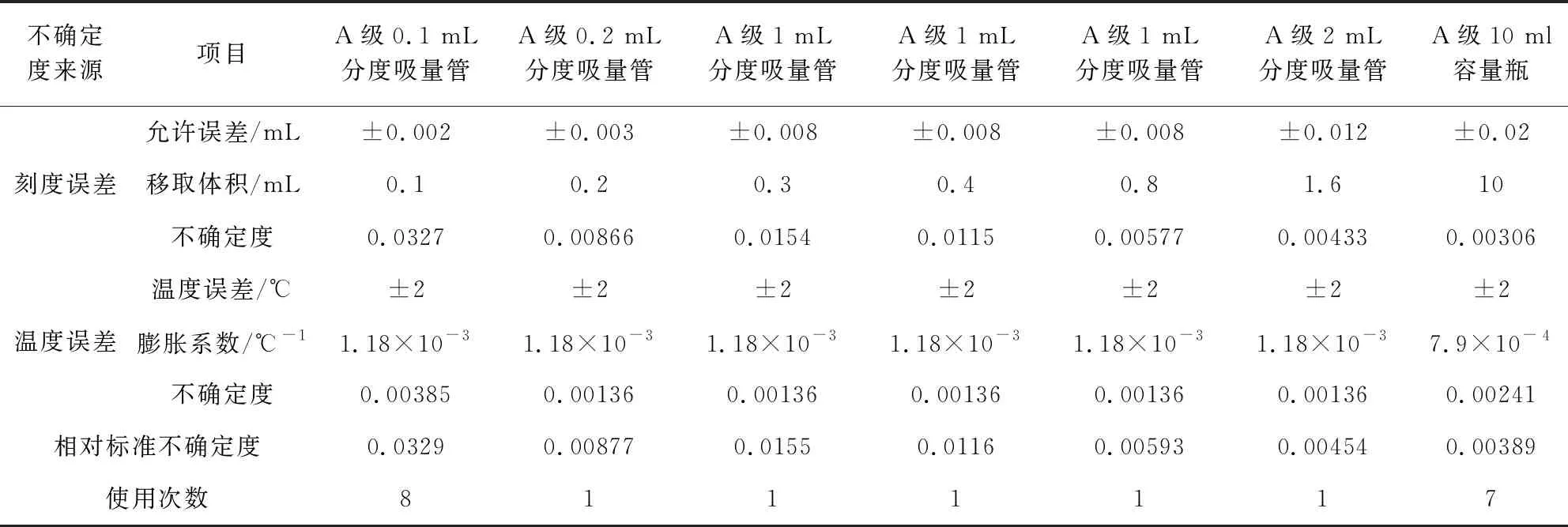
表3 标准曲线配制过程引入的不确定度

⑥标准工作曲线拟合引入的不确定度
本实验对可的松和氢化可的松混标制备了7个不同浓度点的标准工作液,每个浓度点检测3次,工作曲线以外标峰面积/内标峰面积的比值(A/Ai)为纵坐标,标准溶液浓度(Ci)为横坐标,采用最小二乘法拟合,使用权重1/x,得出目标物的回归方程为y=ax+b(其中a为斜率,b为截距)。可的松的线性方程为y=0.0851x+0.0042,氢化可的松线性方程为y=0.1544x+0.0248。标准曲线测定结果见表4。

表4 标准曲线
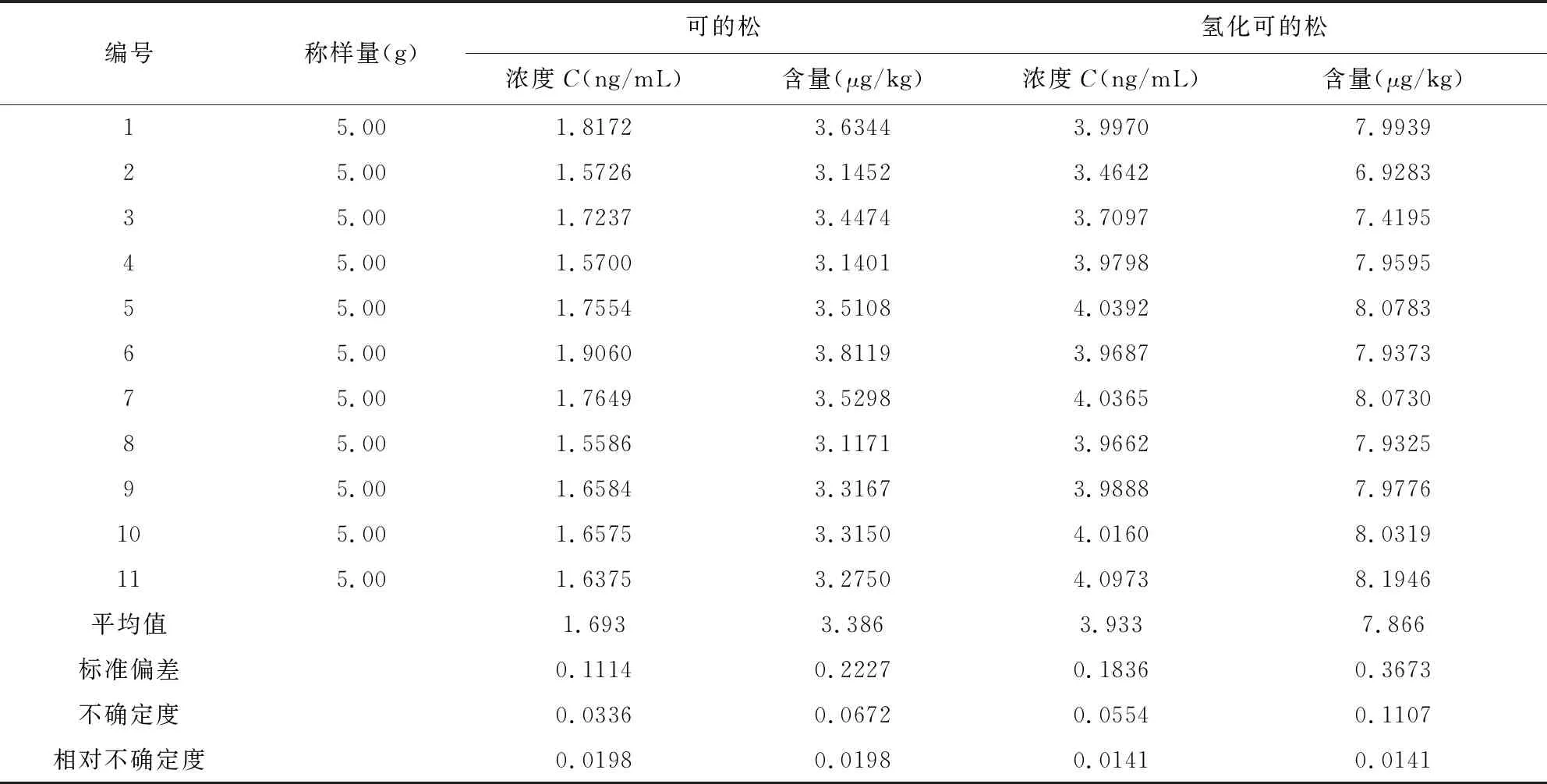
表5 猪肉中可的松和氢化可的松测定结果

Urel(3)=
氢化可的松由标准溶液引入的相对标准不确定度为:

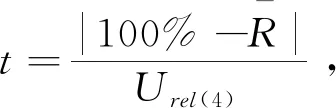

表6 猪肉中可的松和氢化可的松的回收率结果

2.3.6 不确定度的合成和结果表示 样品的称样质量、样品定容、标准物质、测量重复性与回收率等的不确定度分量相互独立,将各不确定度进行合成,即得相对合成不确定度Urel,按下式计算得到可的松:0.0646,氢化可的松:0.0616,所以可的松的合成标准不确定度为0.0646×3.386=0.2187,氢化可的松为0.0616×7.866=0.4845。具体结果见表7。

表7 不确定度分量评定表
2.3.7 扩展不确定度 在置信水平为95%,取包含因子k=2,则可的松和氢化可的松的不确定度分别为U可的松=0.4374、U氢化可的松=0.9690,最终结果可表示为可的松含量X可的松=(3.386±0.4374)μg/kg,氢化可的松含量X氢化可的松=(7.866±0.9690)μg/kg。
3 结 论
本文分析过程中可知可的松和氢化可的松不确定度主要来源于样品回收率、重复性、标准物质及其稀释过程中引入的不确定度,样品回收率涉及到前处理和上机的过程,可能与仪器稳定性以及提取过程中分取提取液人员操作有关系。因此,一是加强人员培训,严格规范操作,以减少人为因素引入的不确定度;二是平时加强仪器的维护保养,检定校准及期间核查,使仪器的各方面指标都处于合理范围之内,减少由于仪器稳定性造成的系统误差;三是在标准工作曲线配制以及前处理过程中,移取溶液时最好使用同一器具及准确度较高的仪器设备,以减小测量的不确定度。
猜你喜欢
制造技术与机床(2017年9期)2017-11-27 02:14:23
电源技术(2015年7期)2015-08-22 08:48:52
橡胶工业(2015年2期)2015-07-29 08:29:46
西南石油大学学报(自然科学版)(2015年3期)2015-04-16 05:12:08
电测与仪表(2015年7期)2015-04-09 11:40:30
西南军医(2014年5期)2014-04-25 07:42:49
电测与仪表(2014年9期)2014-04-15 00:27:16
电测与仪表(2014年11期)2014-04-04 09:21:40
食品工业科技(2014年9期)2014-03-11 18:15:39
电加工与模具(2014年3期)2014-02-24 09:32:27
