
布鲁氏菌生物恐怖战剂RTqPCR现场检测方法研究
2022-12-01 08:06张立安任小侠侯亚欣王慧飞
中国人民警察大学学报 2022年12期
张立安,宗 鹏,任小侠,侯亚欣,王 楠,王慧飞
1.烟台市消防救援支队,山东 烟台 264000; 2.抚州市消防救援支队,江西 抚州 344000; 3.中国兽医药品监察所,北京 100081; 4.中国人民警察大学 a.研究生院; b.侦查学院,河北 廊坊 065000
0 引言
生物恐怖袭击是指利用细菌、病毒和毒素等生物制剂作为武器针对人类、动物和农作物实施攻击,造成伤亡、制造恐慌的一种恐怖主义形式。布鲁氏菌作为失能型生物恐怖战剂,可以感染人和牲畜导致布鲁氏菌病[1-3]。根据《中华人民共和国传染病防治法》,布鲁氏菌病为乙类传染病[4],世界卫生组织将其列为B类法定传染病[5]。根据疾病预防控制局的资料显示,2018年我国布鲁氏菌发病人数为37 947人,截止到2019年11月,2019年发病人数达43 635人。2010年东北农业大学27名学生和1名教师感染了布鲁氏菌病,2019年12月兰州兽医研究所发现96人感染布鲁氏菌病。布鲁氏菌病原体可通过体表皮肤黏膜、消化道、呼吸道侵入机体,人类患病的途径多样,包括食用未经高温消毒的牛奶和奶制品,直接接触受感染的动物组织,误食、吸入或注射培养的布鲁氏菌等[6]。布鲁氏菌病会造成严重的并发症甚至有死亡危险,因此快速精准检测布鲁氏菌、及时进行应急处置显得尤为重要。
自2000年以来,人类布鲁氏菌病是发病数上升速度最快的传染病之一[7-8],而且布鲁氏菌病具有潜伏期,临床分为急性、亚急性、慢性三类,急性潜伏期在3个月以内,慢性潜伏期可达6个月[9]。因此,及时准确侦检是应对布鲁氏菌生物恐怖袭击的有效措施。目前血培养检测布鲁氏菌病是金标准[10-11],我国现行《布鲁氏菌病诊断标准》(WS 269—2019)规定SAT和分离布鲁氏菌均为检测方法。布鲁氏菌传统血清学方法检测的特异性不强,且检测结果受检测人员主观意识影响较大。细菌分离培养技术检测时间较长,检测对试验环境、设备及操作人员要求较高,不适合现场检测。现场试纸检测受检测时机限制及环境温度等影响,其灵敏度检出限还有待技术完善和提高。分子生物学鉴定是近几年逐步发展的技术,具有特异高效的优点,在布鲁氏菌基因检测方面利用核糖体RNA、Omp25、IS711、BCSP31等保守序列做了大量研究和方法构建。实时荧光定量PCR(RTqPCR)方法具有灵敏性高、特异性好等特点,在实验室研究中应用广泛,反恐应急等部门也做了大量尝试和探索[12-14]。1990年,Fekete等开创了布鲁氏菌PCR检测先河,并利用多重PCR进行基因分型[15-17]。本文探讨适合现场快速检测布鲁氏菌的PCR方法,以期提高应急处突检测能力。
1 材料与方法
1.1 试验材料及仪器
试验材料:布鲁氏菌灭活疫苗株(S2),由中国兽医药品监察所惠赠;Sybr Green qPCR mix、Taq PCR MasterMix、细菌基因组DNA快速提取试剂盒均购自北京艾德莱(Aidlab)生物有限公司;D2000 DNA Ladder购自Biosharp公司;Petrifim 6406菌落测试片(3M公司);布鲁氏菌基因的载体构建由中美泰和生物技术(北京)有限公司完成;其他试剂为国产分析纯试剂或其配制而成。主要仪器:LightCycler 1.5型实时荧光定量PCR仪(瑞士Roche公司),MyCycler型PCR仪(美国Bio-rad公司),JY600C型水平电泳仪(北京君意东方电泳设备有限公司),AlphaDigiDoc实时凝胶处理系统(美国Alpha Innotech公司),MSC12型生物安全柜(美国Thermog公司)。
1.2 检测方法
1.2.1 生物信息学分析
利用NCBI公布的16S核糖体RNA基因序列,通过BLAST和DNAMAN 6.0对比分析,利用MEGA 6.06软件的邻接法进行系统进化树构建,分析基因相似性。用Primer Premier 5.0设计上下游引物,如表1所示,引物由中美泰和生物技术(北京)有限公司合成。
表1 引物序列及参数
1.2.2 质粒的提取与鉴定
按照Aidlab高纯质粒小量快速提取试剂盒的操作步骤提取质粒,作为阳性标准品。利用普通PCR进行目的基因鉴定,20 μL反应体系,Taq PCR MasterMix(Aidlab)10 μL,上下游引物B16SF、B16SR各0.5 μL,底物1 μL,双蒸水8 μL。反应程序为:预变性94 ℃,3 min,1个循环;变性94 ℃,10 s;退火/延伸60 ℃,30 s,30个循环。扩增产物进行琼脂糖凝胶电泳后成像鉴定。
1.2.3 荧光定量正交试验
选择退火时间、退火温度、引物量设计3水平3因素如表2所示的L9(34)正交试验。实时荧光定量PCR反应体系为20 μL:Sybr Green qPCR mix 10 μL,上下游引物qBSF1、qBSR2各nμL,底物1 μL,双蒸水8 μL。反应程序为:预变性94 ℃,3 min,1个循环;变性94 ℃,10 s;退火/延伸m℃,ks,40个循环;溶解曲线40~85 ℃,升温速率0.2 ℃·s-1。
表2 参数设置
1.2.4 灵敏度与特异性检测
以10倍梯度稀释得到浓度为10-1~10-7的稀释菌液,取100 μL喷涂在菌落测试片上,37 ℃恒温培养48 h,计数菌斑,计算菌液浓度。使用稀释浓度菌液,按照优化后的反应体系进行RTqPCR反应,Ct值和溶解曲线分析反应灵敏度和特异性。
1.2.5 环境样本测试
采集草坪里的积水和地表土壤样本,样本处理方法为:使用移液枪吸取1 mL采集样本到一个离心管中,3 000 rpm离心5 min,使用移液枪吸取上清液转入新离心管中,13 000 rpm离心30 s,吸取下层液体作为检测样品,进行RTqPCR检测分析。
2 结果与讨论
2.1 重组质粒的鉴定
通过NCBI获得羊、牛、猪等种类布鲁氏菌(Brucella_melitensis_16M、Brucella_abortus、Brucella_ovis、Brucella_suis)的16S rRNA基因序列,通过生物信息学分析,选取高度同源保守序列片段,PCR扩增琼脂糖凝胶电泳获得如图1所示结果,与预期相符,且不在其他位置产生扩增条带。测序对比分析为布鲁氏菌16S rRNA目标基因片段。
图1 PCR扩增凝胶鉴定结果
2.2 正交试验结果与分析
PCR反应过程以单链DNA为模板,在反应溶液中经过变性、退火、延伸、循环使目的基因得以迅速扩增,PCR反应体系主要有Mix、水、引物和样品模板。由于Mix为缓冲体系,具有一定缓冲能力,两步反应中退火过程是引物与模板结合复制的过程,直接影响PCR的反应效率,引物量是一个重要变量。退火温度和退火时间是影响RTqPCR反应的两个条件变量,通过这三个要素的3水平3因素L9(34)正交试验,结果如表3所示。在正交试验中极差R值越大,则表明该因素水平的改变对试验结果影响越大,结果显示引物量的R值为4.68,远大于退火时间的1.93和退火温度的1.62,反应中一般增加反应物浓度有利于反应的正向进行,因此引物量为主要影响因素。k值体现出各单因素对反应的影响。由图2(a)看出随着引物量不断增加,Ct值呈递减趋势,0.8 μL时最低,说明随着引物量的增加,反应进入指数反应越早,反应越快。但同时也要考虑成本等其他因素,PCR是一个典型的酶促反应,选择反应体系的最适温度显然有利于反应的进行。从图2(b)看出随着退火温度升高,Ct值呈现先增后降趋势,59 ℃时最低,60 ℃时最高,表明在59 ℃时更有利于反应的进行。图2(c)则随着退火时间的增加,Ct值先抑后扬,30 s时Ct值最低,说明退火时间过短和过长都不利反应的进行。因此,通过正交试验确定三个要素的最优反应条件为A3B1C2,最终确定的最优反应条件为:总反应体系为20 μL,包括Sybr Green qPCR mix 10 μL,上下游引物各0.8 μL,超纯水7.4 μL,引物1 μL;采用两步法预变性94 ℃持续5 min,变性94 ℃持续10 s,退火延伸59 ℃持续30 s;扩增40个循环,扩增阶段温度变化速率20 ℃·s-1,每次在退火结束后收集荧光信号;溶解阶段从40 ℃向85 ℃升温溶解,升温速率0.2 ℃·s-1,持续收集荧光信号。
表3 正交反应结果分析
2.3 实时荧光定量PCR灵敏度与特异性分析
2.3.1 菌落的确定
细菌在Petrifilm测试片上生长时,细胞代谢产物与上层指示剂氯化三苯基四氮唑(TTC)发生氧化还原反应,将指示剂还原成红色非溶解性产物三苯甲,从而使细菌着色,故测试片上红色菌落判断为菌落总数,测试片菌落计数适宜范围为25~250 CFU。若所有稀释度的平均菌落数均不在区间,则以最接近区间的平均菌落数乘以稀释倍数计算。将培养的菌液稀释10-1~10-7倍,喷涂于菌落测试膜片上,37 ℃恒温培养48 h,结果如图3所示,稀释10-5倍的菌液在菌落测试片上形成菌斑远超250个,稀释10-7倍的菌液在测试片上几乎看不到菌落,稀释10-6倍的菌液在测试片上形成菌斑平均为22个,其菌落数量处理如表4所示,原始菌落浓度为2.2×108CFU·mL-1。
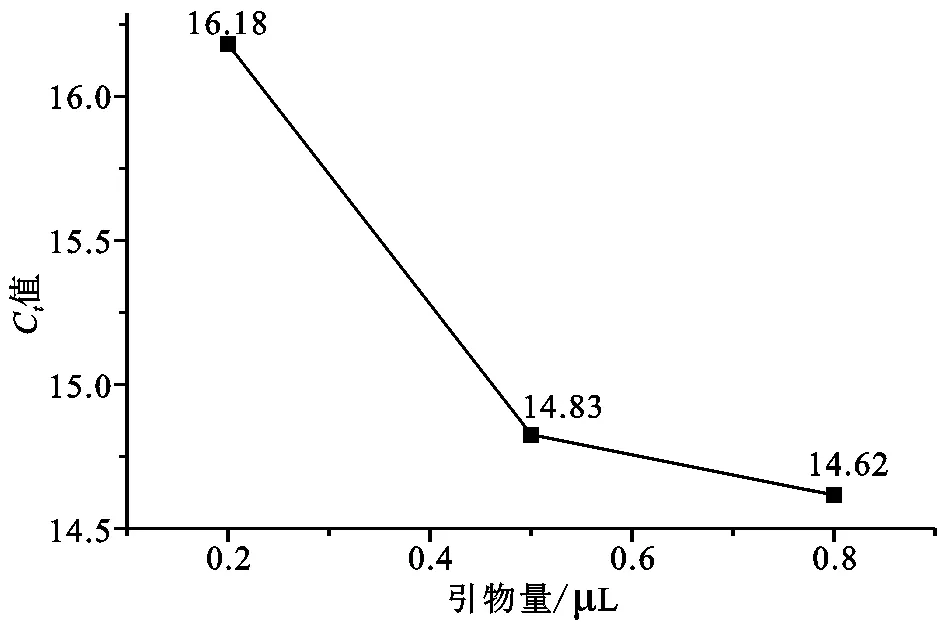
(a)引物量
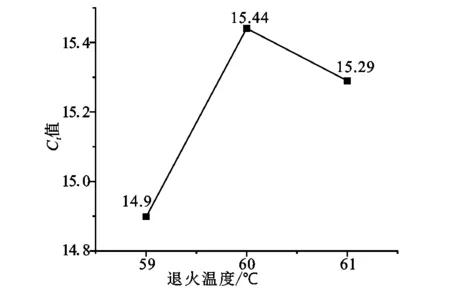
(b)退火温度
(c)退火时间
2.3.2 RTqPCR灵敏性分析
RTqPCR是利用荧光信号监测PCR过程,只要有荧光信号便可以监测出扩增过程。根据菌落计数,进行菌液浓度梯度实时荧光定量PCR检测,图4中1号样为阳性对照,2~6号样分别为菌液稀释10-3、10-4、10-5、10-6和10-7倍浓度,7号样为阴性对照。表5列出各组Ct值,随着稀释倍数的增加,Ct值不断增大,5号样本菌液稀释10-6倍时Ct值平均为25.3±0.057,6号样本菌液稀释10-7倍时Ct值平均为30.3±0.027。由于采用荧光染料法,实时荧光信号曲线的Ct值在30个循环后可能出现非特异性扩增,且菌液稀释10-7倍时菌落无法计数,因此RTqPCR检测的菌液浓度至少为220 CFU·mL-1。
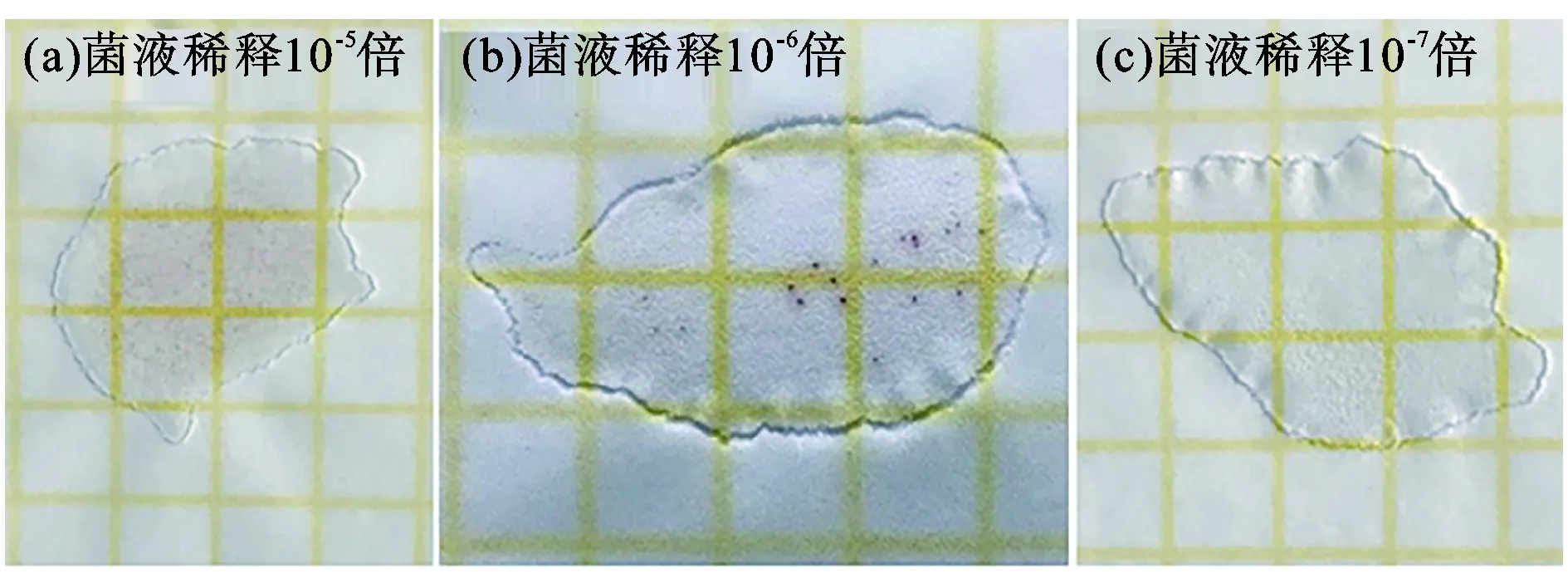
图3 不同稀释倍数菌液测试片培养结果

表4 菌落计数统计表
表5 不同稀释浓度布鲁氏菌RTqPCR扩增Ct值
图4 不同稀释浓度布鲁氏菌RTqPCR反应
2.3.3 RTqPCR特异性分析
利用RTqPCR溶解曲线来检验扩增产物的单一性,理想的特异性PCR反应一对引物只生成一种产物,且溶解曲线峰值只有一个,图5(a)阳性样本和布鲁氏菌疫苗株S2反应迅速,很快进入指数反应阶段,呈现S形荧光扩增曲线,其Ct值分别为9.8和13.6;阴性对照和大肠杆菌检测结果均为一条平滑直线,均无荧光值增长。图5(b)显示在65 ℃附近,出现明显单一峰,没有出现双峰、叠峰、多峰等现象,说明RTqPCR反应没有出现引物二聚体和非特异性扩增。表明引物设计具有非常强的特异性,扩增产物单一,利用16S rRNA基因片段作为检测鉴定布鲁氏菌具有相当可靠性。
2.4 环境样本测试
通过采集室外草坪里的积水和土壤样本,样本处理后,进行RTqPCR检测,结果如图6所示,阳性对照产生特异性扩增,土壤样本和水体样本Ct值在30以内没有出现扩增信号,表明目标检测区域没有阳性样本存在。

图5 RTqPCR特异性检测图
3 结论
根据NCBI公布的16S核糖体RNA基因保守区域序列设计合成特异性引物,通过PCR反应进行目的基因的扩增,构建并鉴定重组质粒,优化反应条件和扩增体系。用于检测的特异性目的片段16S rDNA是一种常用的布鲁氏菌PCR检测目标,扩增片段在布鲁氏菌属DNA中高度保守,可以有效将布鲁氏菌与其他细菌分离,有利于荧光定量PCR检测的应用。检测样本经简单处理直接作为PCR检测底物,省去了核酸提取步骤,适用于从各种相态环境样本中快速筛选存在布鲁氏菌基因组DNA的样品。得到本方法用于样品检测的检测限为220 CFU·mL-1菌浓度,并且体系有助于反应快速进入指数反应,Ct值出现早,且表现出良好特异性。通过采集自然水样和土壤样本进行环境样本实际检测,结果表明该检测方法对布鲁氏菌具有特异性,环境中其他菌株不会对检测结果造成影响。可见,在执行生物恐怖袭击现场侦检任务时,基于实时荧光定量PCR技术的基因检测方法能对布鲁氏菌快速、准确识别,满足检测需求,为生物恐怖袭击现场快速检测布鲁氏菌提供一种实用检测手段。
图6 模拟检测荧光信号图
猜你喜欢
中国土壤与肥料(2021年5期)2021-12-02
复旦学报(医学版)(2021年4期)2021-08-05
现代畜牧科技(2021年4期)2021-07-21
疯狂英语·新悦读(2020年7期)2020-07-30
癌变·畸变·突变(2020年1期)2020-02-12
中国民族医药杂志(2016年9期)2016-05-09
现代食品(2016年24期)2016-04-28
上海蔬菜(2015年2期)2015-12-26
食品工程(2015年3期)2015-12-07
电测与仪表(2015年20期)2015-04-09
