
骨巨细胞瘤发病现状及驱动RANKL/OPG失衡机制的研究进展*
2022-11-30 15:15:20景豆豆郭昊宇浦飞飞邵增务
华中科技大学学报(医学版) 2022年5期
曹 理, 吴 蔚, 景豆豆, 郭昊宇, 浦飞飞, 邵增务
华中科技大学同济医学院附属协和医院骨科,武汉 430022
1 骨巨细胞瘤的发病现状
1.1 流行病学特点
世界范围内原发的骨巨细胞瘤(giant cell tumor of bone,GCTB)占所有骨肿瘤的4%~5%,但我国骨肿瘤患者中这一比例明显偏高,可达13%~20%,发病高峰出现在30岁前后,男女比例为1.4∶1,20岁至40岁的患病人群占总患病人群的70%以上[1-4]。一般而言,90%的GCTB好发于四肢长骨干骺端,如股骨远端(32%~37%)、胫骨近端(25%~28%)、桡骨远端(10%~11%)、股骨近端(7%~8%)和腓骨近端(5%~8%)等部位,随疾病进展常侵犯骨骺部[5-6]。10%的GCTB则见于短骨、扁骨或某些不规则骨,如骶骨(5%~9%)、胸腰椎骨(2%~3%)和手足(1%~2%)等部位[7-8]。少部分GCTB可继发于Paget骨病(Paget’s disease of bone,PDB),发生率约1%,简称PDB/GCT[9]。其在意大利和英国的白人中报道较多,恶性程度较高,发病高峰在40岁以后,男性继发率高于女性,5年生存率低于50%,好发于骨盆、脊柱和颌面部不规则骨,具有家族聚集效应[10-11]。若PDB/GCT患者出现多发性骨巨细胞瘤病灶,则以中轴骨上的病灶数量居多[10]。
1.2 临床转归特点
GCTB作为交界性肿瘤,死亡率低,总体恶性率为5%~10%[12],但存在发病隐匿、局部复发、恶性转化和肺转移等不良后果,给患者带来不必要的痛苦和经济负担。
GCTB发病速度快且临床症状隐匿,常表现为局部疼痛和偶发的关节活动受限,易被忽视,5%~30%的患者患病后可无任何症状,常因在外伤或骨骼肌牵拉后出现病理性骨折而就诊[13-15]。少部分GCTB原发于脊柱、骶骨等部位,由于局部压迫或压缩性骨折,可出现下腰痛、下肢放射痛、大小便功能障碍或性功能障碍等神经压迫症状[15]。而PDB/GCT患者发病则以头面部、脊柱和骨盆为主[16-17]。出现在头面部蝶骨或颞骨的病灶往往更具侵袭性,可致毗邻的神经受压而出现头痛、吞咽困难、外展神经麻痹和复视等症状[18]。
GCTB局部复发率高,常采用的病灶刮除手术治疗后2年的复发率可达25%~65%[19-20]。一些小样本研究报道在手术中辅以液氮的冷冻消融、高速磨钻、双氧水、苯酚或氯化锌浸渍等方法,中位复发率最低能降至11%[21]。地诺单抗和双磷酸盐类药物可作为手术前后的辅助治疗预防复发,但目前最佳用药方案尚缺乏高质量的临床研究数据支持[20,22]。GCTB刮除手术虽然可通过在骨皮质开窗刮除病灶清除大量肿瘤细胞,但也会造成髓腔内微血管破裂和周围软组织破坏,从而导致医源性肿瘤播散,在边界处形成新的肿瘤微小病灶,使GCTB在复发时病灶增殖侵袭速度更快,可能需要二次扩大刮除或骨骼切除重建手术,给患者带来经济负担和机体功能障碍[23]。
GCTB存在恶性转化的风险且预后差。1.6%的GCTB为原发恶性,而3%~8%的GCTB在放疗、手术、靶向治疗等情况下继发恶性转化[4]。恶性转化的GCTB组织中可观察到低分化的多形性单核细胞结节,具有高级别肉瘤表现[24-26]。或在典型的GCTB细胞背景中,出现恶性转化的骨肉瘤、低分化多形性肉瘤或纤维肉瘤病灶[27-28]。Mayo诊所的数据表明接受过放射治疗的GCTB患者中,76%的人群随访10年后出现了继发性的骨巨细胞瘤恶性转化,其中67%~80%恶性转化的GCTB曾接受过放射治疗,且发病年龄偏高[29-30]。GCTB出现恶性转化的人群中,68岁以上、39~68岁和39岁以下人群的5年生存率分别为1%、33%和70%[24]。恶性转化机制可能与单核梭形基质细胞RUNX2转录因子(runt related transcription factor 2)高表达、mTOR/PI3K/AKT(mechanistic target of rapamycin kinase/phosphatidylinositol 3 kinase/protein kinase B)通路活化和分泌过量的趋化因子CCL20(C-C motif chemokine ligand 20,CCL20)等因素有关[31-33]。
GCTB存在肺转移的风险,原发性和复发性GCTB转移率分别为3.2%和9.8%[34]。GCTB肺转移的机制可能与GCTB细胞通过局部侵袭血管或在病理性骨折的影响下形成肿瘤内血栓,进而血性转移至肺部有关[15,35]。但GCTB的肺转移灶生长缓慢,转移结节小于5 mm的患者中5年无进展生存率达到60%,而大于5 mm者中则为25%[36]。Rizzoli研究中心回顾649例GCTB患者,提示出现肺部转移的平均时间为35.2个月[37]。Ebeid等[34]回顾15名肺转移的GCTB患者,有1例死于多发肺转移的并发症。Jidveian等[38]认为GCTB复发和肺转移风险将给患者带来心理压力,可能导致焦虑或抑郁情绪。
1.3 发病机制
多核巨细胞过度激活是GCTB骨质破坏的直接原因。光镜下可观察到GCTB大量的反应性多核巨细胞,夹杂着少量单核巨噬细胞和单核梭形基质细胞[39]。其中多核巨细胞核数目通常达40~50个,胞内富含嗜酸性的细胞质和液泡[40]。这些多核巨细胞占据骨巨细胞瘤瘤体50%以上体积,通过抗酒石酸酸性磷酸酶(tartrate-resistant acid phosphatase,TRAP)、基质金属蛋白酶2(matrix metallopeptidase 2,MMP2)、基质金属蛋白酶9(matrix metallopeptidase 9,MMP9)和组织蛋白酶(cathepsin K,CTSK)途径介导GCTB溶骨性骨质破坏、假囊性变和肿瘤中心变性坏死[35,41]。
单核梭形基质细胞恶性增殖是GCTB持续进展的根本原因。首先,瘤体中的单核梭形基质细胞作为成骨细胞的前体本身增殖潜能强,细胞内的基因突变和染色体端粒不稳改变了表观遗传调控稳态,最终带来基质细胞增殖堆积、成骨分化障碍和骨代谢细胞因子RANKL/OPG平衡失调,诱发GCTB恶性转化[42-44]。其次,研究发现异种移植模型中的单核梭形基质细胞,可在体外独立培养增殖,表达增殖标记鼠双微体2同源体MDM2(mouse double minute 2 homolog)和Ki67(marker of proliferation Ki-67),并在动物模型内独立生长成瘤,通过旁分泌CCL20诱导骨髓单核细胞发展为多核巨细胞[31,45-46]。最后,单核梭形基质细胞通过分泌SDF-1趋化因子(stromal cell-derived factor 1)和SRGN糖蛋白(serglycin),以血管内皮迁移的方式募集血液中单核细胞至GCTB病灶并诱导其分化为多核巨细胞[47-48]。所以,单核梭形基质细胞被认为是GCTB的“罪魁祸首”,其恶性增殖与GCTB的侵袭、恶性转化和复发紧密相关[49]。类似的,继发的PDB/GCT同样存在ZNF687基因始祖突变所带来的转录稳态失调,单核梭形基质细胞恶性增殖的影响被放大,出现更为严重的骨小梁结构异常、骨质破坏和紊乱的反应性成骨。同时有研究报道85%的PDB/GCT病灶组织内可观察到病毒样包涵体,提示病毒可能参与到PDB/GCT的进展[10,50-52]。
RANKL/OPG平衡失调是GCTB病变的核心环节。生理条件下,骨骼微环境中来自成骨细胞系的单核梭形基质细胞在细胞膜表达适量的RANKL,与骨髓造血系的破骨细胞前体细胞膜上的RANK(receptor activator of nuclear factor kappa B)相互作用,通过胞内PI3K/Akt途径、IKKs/NF-κB(I-kappa B kinases/nuclear factor kappa B)途径和JNK/MAPK/c-Jun(c-Jun N-terminal kinase/mitogen-activated protein kinase/cellular Jun)途径促进破骨细胞存活并融合形成多核巨细胞发挥溶骨功能,而成骨细胞及其前体分泌的诱饵受体OPG通过竞争性结合RANKL阻断这一效应,从而避免过度的骨质溶解[53-54]。GCTB条件下,异常的驱动因素导致骨骼微环境RANKL/OPG原有平衡被打破。单核梭形基质细胞不仅高表达RANKL促进多核巨细胞侵袭性溶骨,而且通过RANKL-RANK系统大量分泌白介素(interleukin,IL)-1β、IL-6、IL-8、IL-11、IL-17、巨噬细胞集落刺激因子(macrophage colony-stimulating factor,M-CSF)、肿瘤坏死因子(tumor necrosis factor-α,TNF-α)、甲状旁腺激素相关蛋白(parathyroid hormone-releasing protein,PTHrP)和前列腺素E(prostaglandin E),扰乱成骨细胞分泌降低了的OPG水平,进一步加剧RANKL/OPG比例升高[53,55-56]。GCTB病灶内破骨细胞和多核巨细胞数量逐渐占据优势,通过侵袭溶骨“挤压”成骨细胞生存空间,最终增加骨破坏和恶性转化,加速GCTB进展[53,55,57-58]。针对RANKL开发的单克隆抗体如地诺单抗,在临床展现出良好疗效,已成为GCTB辅助治疗的一线药物[20]。然而,既往研究关注点集中于RANKL/RANK/OPG下游效应,近年来RANKL/OPG平衡失调的异常驱动因素逐渐被报道,明确驱动因素及其互作分子将有助于深入理解GCTB发病机制,为药物研制提供新的靶点。
2 骨巨细胞瘤RANKL/OPG平衡失调驱动因素分析
2.1 H3F3A基因突变经由表观遗传学调控驱动RANKL/OPG平衡失调
H3F3A基因位于1号染色体q42.12区,与17号染色体的H3F3B基因一同编码H3组蛋白的变体——H3.3组蛋白[4,59]。不同于H3.1和H3.2组蛋白,H3.3组蛋白以不依赖DNA复制的方式,在伴侣分子HIRA(histone cell cycle regulator)的协助下沉积至核小体高频更替区域的转录位点[60]。此外,H3.3组蛋白游离的N端存在许多翻译后修饰位点(post-translational modifications,PTMs),如H3.3K9me1、H3.3K27me3、H3.3K36me3和H3.3K14ac等,异常的PTMs将显著改变H3.3组蛋白生理功能,激活或抑制基因转录,演变为驱动GCTB的“癌组蛋白”[61-62](图1)。
GCTB组织活检发现94.3%~100%的患者存在H3F3A基因突变,导致H3.3组蛋白上34位甘氨酸(G34)变为色氨酸(G34W),或更为罕见的亮氨酸(G34L)、缬氨酸(G34R)和甲硫氨酸(G34R)等[63-66]。G34作为甲基转移酶SETD2(SET domain containing 2)和PRC2(polycomb repressive complex 2)结合H3.3组蛋白的关键位点,突变后的氨基酸由于空间位阻的变化显著地改变了SETD2和PRC2酶活性,进而通过顺式作用改变G34两侧的K36和K27的甲基化稳态[67-68]。H3.3K36在增强子区域甲基化水平较高,被视为转录抑制标记;而H3.3K27可成为转录延伸阶段的甲基化标记位点,进而募集其他转录因子广泛地影响转录[62,69]。一方面,SETD2介导的K36甲基化降低通过抑制成骨相关基因转录使得GCTB单核梭形基质细胞成骨能力下降,维持在低度分化的增殖状态[70]。另一方面,PRC2介导的部分增强子区域K27甲基化水平升高导致部分转录因子(如OPG基因转录因子E2F)失活,下游分泌破骨细胞生成抑制因子OPG减少,RANKL/OPG比例升高,GCTB单核梭形基质细胞诱导破骨细胞分化和激活的能力增强,最终导致溶骨性破坏[71]。
2.2 ZNF687基因突变经由转录枢纽调控驱动RANKL/OPG平衡失调
ZNF687基因编码具有C2H2锌指结构域的转录枢纽蛋白,在肝癌、白血病和肺腺癌中具有促癌功能[72-74]。PDB/GCT家族队列和PDB/GCT散发人群中,ZNF687基因2810位C突变为G(c.2810C>G)的发生率为67%~86%,进而导致ZNF687蛋白P937R的错义突变[52]。在美国黑人PDB/GCT患者中同样发现ZNF687蛋白相同的P937R突变,证实不同基因背景下ZNF687基因突变与PDB/GCT存在普遍性关联[75]。
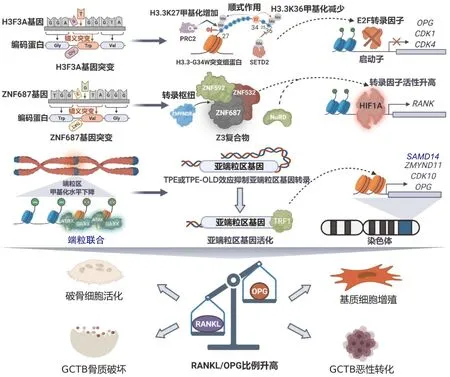
图1 骨巨细胞瘤发病的驱动因素Fig.1 Driving factors in giant cell tumor of bone
ZNF687蛋白突变后,同ZNF592蛋白(zinc finger protein 592)和ZNF532蛋白(zinc finger protein 532)构成的异源三聚体Z3复合物活性异常升高,通过复合物NuRD(nucleosome remodeling and histone deacetylase)、转录调节因子ZMYND8(zinc finger MYND-type containing 8)或转录因子HIF1A(hypoxia inducible factor 1 subunit alpha)等促进PDB/GCT进展[76-77]。首先是破骨细胞活性受到影响,携带ZNF687突变的PDB/GCT患者破骨细胞内RANK表达升高,可耗竭OPG的负调节效应,破骨细胞前体进入到破骨细胞之间的融合阶段启动溶骨,融合后的破骨细胞核数目可达153个,大量骨质破坏释放的细胞因子给予了PDB伴发GCT的土壤[75,78]。另一方面,突变后的Z3复合物失去经由ZMYND8因子特异性标记组蛋白乙酰化发挥修复DNA的功能,DNA损伤后ZMYND8因子不再形成NuRD-GATA2A复合物(nucleosome remodeling and histone deacetylase-GATA binding protein 2)维持DNA损伤端的稳态,导致单核梭形基质细胞加速DNA修复和异常增殖,表达大量RANKL进一步刺激破骨细胞溶解骨质[79-80]。ZNF687基因突变引起破骨细胞与单核梭形基质细胞形成“溶骨-增殖-再溶骨”的恶性循环,成为PDB/GCT发病的重要机制。
2.3 端粒联合通过染色体末端调控驱动RANKL/OPG平衡失调
正常体细胞端粒长度随着细胞复制而不断缩短,较长的端粒通过端粒位置效应(telomere position effect,TPE)或长距离端粒位置效应(telomere position effect-over long distances,TPE-OLD)抑制亚端粒区基因表达,一旦端粒随细胞复制缩短将启动细胞凋亡以防止癌变[81]。70%~85%的GCTB患者中存在端粒缩短后通过异源染色体两端端粒互相结合抵抗细胞凋亡的现象,称为端粒联合(telomeric associations,TAs)[82-83]。
TAs一方面通过破坏基因组完整性,抑制端粒形成保护性“D”环或“T”环结构,释放DNA损伤信号加速单核梭形基质细胞DNA异常修复;另一方面影响端粒通过组蛋白调控的染色体末端基因表达,改变亚端粒区基因表达模式抵抗凋亡[83-84]。与正常细胞相比,携带H3.3-G34W突变的GCTB患者染色质甲基化整体水平下降20%,两种伴侣蛋白ATRX(alpha thalassemia/mental retardation syndrome x-linked)和DAXX(death domain associated protein)协助H3.3组蛋白沉积在端粒区使之甲基化水平下降,端粒结合蛋白TRF1(telomeric repeat binding factor 1)对端粒亲和力升高,TAs发生率显著升高[85-86]。端粒区域由于TAs带来的基因组不稳、TPE和TPE-OLD的调控失衡,使得单核梭形基质细胞的错译突变通过组蛋白不断地被“放大”,导致亚端粒区的基因表达受到影响[87]。SAMD14(sterile alpha motif domain containing 14)、ZMYND11(zinc finger MYND-type containing 11)、CDK10(cyclin dependent kinase 10)和OPG等调控基质细胞增殖分化和诱导破骨细胞形成的基因均位于亚端粒区,可能受到TAs的影响从而降低其表达水平,驱动RANKL/OPG升高,加速单核梭形基质细胞增殖,诱导破骨细胞形成,演变为GCTB[86]。
3 展望
通过高通量测序技术,研究者在过去10年间发现原发和继发的GCTB中广泛存在的基因突变,在机制层面向我们展示了GCTB内RANKL/OPG失衡的驱动因素。本文综述了GCTB发病的分子机制特点和基因突变驱动GCTB的重要研究进展,总结了突变所带来的下游关键分子的变化趋势,为GCTB诊断治疗提供新的思路。目前RANKL/OPG作为临床治疗GCTB的重要靶点效果良好,继续在发病机制层面深入研究将为精确诊断骨巨细胞瘤、开发单克隆抗体药物和开展GCTB遗传学诊断提供新的思路。针对基因突变所驱动的GCTB带来的下游效应仍需更多的探索,这将是有效治疗骨巨细胞瘤的基础。
猜你喜欢
中国药学药品知识仓库(2022年8期)2022-05-09 13:54:24
癌变·畸变·突变(2019年3期)2019-06-03 06:13:04
西安建筑科技大学学报(自然科学版)(2016年5期)2016-11-10 02:39:26
健康管理(2016年2期)2016-05-30 21:36:03
恋爱婚姻家庭·养生版(2016年5期)2016-05-06 20:18:48
分析测试学报(2015年8期)2016-01-13 06:19:36
首都医科大学学报(2015年4期)2015-12-16 13:00:08
实用手外科杂志(2015年3期)2015-08-27 01:53:06
无机化学学报(2014年12期)2014-02-28 17:33:53
无机化学学报(2014年7期)2014-02-28 17:32:11
