
Sequencing of the complete mitochondrial genomes of Carbula crassiventris and Carpocoris purpureipennis and phylogenetic analysis of Pentatominae (Hemiptera:Pentatomidae)
2022-11-28 07:36LIRongRongLIMinSUNShanShanYANJiangZHANGHuFangBAIMing
昆虫学报 2022年10期
LI Rong-Rong,LI Min,SUN Shan-Shan,YAN Jiang,ZHANG Hu-Fang,BAI Ming
(1.Taiyuan Normal University,Jinzhong,Shanxi 030619,China;2.Xinzhou Teachers University,Xinzhou,Shanxi 034000,China;3.Key Laboratory of Zoological Systematics and Evolution,Institute of Zoology,Chinese Academy of Sciences,Beijing 100101,China)
Abstract:【Aim】 In this study,the complete mitochondrial genomes (mtgenomes) of Carbula crassiventris and Carpocoris purpureipennis were sequenced to investigate the mitogenomic characteristics and reconstruct the phylogenetic relationships of Pentatominae.【Methods】 The complete mtgenomes of Carbula crassiventris and Carpocoris purpureipennis were sequenced using the Illumina MiSeq Platform,and the resulted sequences were assembled and annotated.The phylogenetic trees of Pentatominae were reconstructed using Bayesian inference (BI) and maximum likelihood (ML) methods based on the sequences of the 1st and 2nd positions of the codons of the 13 protein-coding genes (PCGs) and the nucleotide sequences of the two rRNA genes of the mtgenome of the two species and other 30 Pentatominae taxa.【Results】 The mtgenome of Carbula crassiventris and Carpocoris purpureipennis are 15 824 and 16 575 bp in length,respectively,with 13 PCGs,2 rRNA genes,22 tRNA genes,and a control region.The mtgenome gene arrangements are conserved within Pentatominae and no rearrangement was detected.In addition,the base composition,codon usage and RNA structures are conserved within Pentatominae.The length,types and copies of repeat units in control regions are different among species.The phylogenetic tree based on BI and ML showed a stable clade comprised of Eysarcorini,Carpocorini,Nezarini and Antestiini.【Conclusion】 The phylogenetic analysis confirmed that Carbula belongs to Eysarcorini,and Carpocoris,Dolycoris and Rubiconia belong to Carpocorini.
Key words:Pentatominae;Carbula crassiventris;Carpocoris purpureipennis;mtgenome;phylogeny
1 INTRODUCTION
Pentatominae is the most chaotic subfamily of Pentatomidae (Rideretal.,2018).Members of this subfamily occur all over the world and occupy terrestrial habitats.Most species are phytophagous and can injure plants by sucking fluid sap from leaves,immature stems,and inflorescences.Pentatominae is the only subfamily of Pentatomidae that can not be well-defined by unique apomorphies,and the classification of Pentatominae varies from worker to worker;furthermore,the phylogenetic relationships of tribes and genera also remain controversial (Graziaetal.,2008;Yuanetal.,2015;Rideretal.,2018).Carpocorini and Eysarcorini are two tribes of Pentatominae that are difficult to be characterized.Variation in structure and color within both of the tribes are obvious,but ambiguous when compared to other tribes.The genera of Carpocorini such asRubiconia,Dolycorisand even the type genusCarpocoriswere once transferred to other tribes of Pentatominae (Puton,1869;Yang,1962).Another member,Aulacetrus,was once considered as a valid tribe named Aulacetrini,and its validation is still under controversial.Similar problems were also found in Eysarcorini.For example,Carbulawas placed in the tribe Palomenini,theCarbulagroup,and Eyarcorini successively (Yang,1962;Linnavuori,1982;Rider,2006).In addition,there are little distinctive characteristics to separate Eysarcorini from the Carpocorini,except for those few genera in which the scutellum is enlarged (Rideretal.,2018).Species of Eysarcorini usually have an expanded and spatulate scutellum with the exception ofCarbula,AspaviaandDurmia.There are some carpocorine genera (e.g.,RubiconiaandCoenus) that also have an expanded,and spatulate scutellum.
The insect mitochondrial genome (mtgenome) is typically a circular,double-stranded DNA molecule,15-18 kb in length,and encodes 37 genes:13 protein-coding genes (PCGs),two rRNA genes,22 tRNA genes,and a control region (also known as the A+T-rich region) that is essential for transcription and replication (Boore,1999;Cameron and Whiting,2008;Françosoetal.,2016).The insect mtgenome has been widely used in species classification,population genetic structure,evolution biology,phylogenetic and biogeographic studies due to its relatively small size,haploid nature,high rate of evolution,relatively conserved gene content,and organization (Wolstenholme,1992;Simonetal.,2006;Maetal.,2012;Cameron,2014;Zhuetal.,2017;Liuetal.,2019).
Until now,only 30 complete or nearly complete Pentatominae mtgenomes have been sequenced (GenBank,June 18,2021),and this is still quite restricted to clarify the phylogenetic relationships of tribes and genera of Pentatominae.In this study,the mtgenomes ofCarbulacrassiventrisandCarpocorispurpureipenniswere sequenced and compared with previously sequenced Pentatominae mtgenomes.Furthermore,the phylogenetic relationships of Pentatominae were reconstructed using Bayesian inference (BI) and maximum likelihood (ML) methods.This study will enhance our understanding of the molecular evolution and phylogenetic relationships among tribes and genera within Pentatominae.
2 MATERIALS AND METHODS
2.1 Sample collection and mtgenome sequencing
Specimens ofCarbulacrassiventrisandCarpocorispurpureipenniswere collected from fields in Dali,Yunnan,and Lvliang,Shanxi,China,on 15 August and 2 June 2015,respectively.The collected samples were immediately impregnated in absolute ethyl alcohol and preserved at -20℃.The Genomic DNA Extraction Kit (BS88504;Sangon,Shanghai) was used to extract genomic DNA from the leg muscles of a single specimen.The mtgenomes were then sequenced on the Illumina MiSeq Platform (Personalbio,Shanghai).FastQC (https:∥www.bioinformatics.babraham.ac.uk/projects/fastqc/;12 October 2021) was used to ensure sequence quality.The fastp v0.20.0 software (Chenetal.,2018) was used to remove adapter sequences and low-quality sequences (q-value<20,length<50 bp),and a total of 19 190 236 and 18 768 248 paired-end clean reads were obtained from 23 215 218 and 22 559 008 raw data,respectively.Then the clean reads were assembled based on A5-miseq v20150522 (Coiletal.,2015) and SPAdes v3.9.0 (Bankevichetal.,2012).
2.2 Mtgenome annotation and sequence analysis
The newly sequenced mtgenomes ofCarbulacrassiventrisandCarpocorispurpureipenniswere annotated using Geneious Prime v9.1.4 software (Kearseetal.,2012).NCBI’s open reading frame finder (ORF finder)(http:∥www.ncbi.nlm.nih.gov/orf/gorf.html) was applied to identify the boundaries of 13 PCGs using the invertebrate mitochondrial code.The 22 tRNA genes were identified using the MITOS web server (Berntetal.,2013).The boundaries of two rRNA genes were confirmed by alignment with previously sequenced mitochondrial sequences.The location of the control region was confirmed by the boundary of the neighboring genes.
MEGA-X was used to analyze the nucleotide composition,codon usage,and amino acid composition (Kumaretal.,2013).Strand asymmetry was calculated using the following calculation formula:AT-skew=(A-T)/(A+T) and GC-skew=(G-C)/(G+C)(Perna and Kocher,1995).The evolutionary rate analysis and a sliding window analysis (a sliding window of 100 bp;step size of 25) were conducted with DnaSP v5.0 based on 13 aligned PCGs (Rozasetal.,2009).Genetic distances between species were estimated using MEGA-X with Kimura-2-parameter based on each PCG.The Tandem Repeats Finder webserver was used to predict tandem repeats in control regions (Benson,1999).
2.3 Phylogenetic analyses
The newly sequenced mtgenomes ofCarbulacrassiventrisandCarpocorispurpureipennisas well as those of 30 Pentatominae taxa,and those of two Asopinae species (used as the outgroup) were used to conduct the phylogenetic analyses.The sequences of the 13 PCGs and two rRNA genes were extracted by Phylosuite v1.2.2 (Zhangetal.,2020).Alignment of PCGs and rRNA genes was conducted using MAFFT v7.475 (Katoh and Standley,2013) according to codon-based and normal alignment models,respectively.The nucleotide substitution saturation of PCGs was measured by DAMBE (Xia,2018),and the database of the 1st and 2nd positions of the codons of the 13 PCGs was extracted by MEGA-X.All alignments were concatenated into a single data matrix using the concatenate sequence function embedded in Phylosuite v1.2.2.Mrbayes v3.2.7a (Ronquistetal.,2012) and IQ-TREE web server (Trifinopoulosetal.,2016) were used to reconstruct the phylogenetic tree using BI and ML methods,respectively.For the BI method,the best-fit partitioning model,GTR+F+I+G4,was selected by ModelFinder installed in Phylosuite v1.2.2 (Kalyaanamoorthyetal.,2017).Four independent Markov chains (three heated and one cold) were run for 10 000 000 generations and trees were sampled every 1 000 generations.The first 25% of samples were discarded as burn-in when the average standard deviation of split frequencies was <0.01.For the ML method,the best-fit partitioning model,GTR+F+I+G4,was selected by model selection in the IQ-TREE web server (Kalyaanamoorthyetal.,2017),and the node confidence was assessed with 1 000 ultrafast bootstrap replications.
3 RESULTS
3.1 Mtgenome structure
The newly sequenced complete mtgenomes ofCarbulacrassiventrisandCarpocorispurpureipennisare 15 824 and 16 575 bp in length,respectively,encoding a complete set of 37 genes including 13 PCGs,2 rRNA genes,22 tRNA genes,and a non-coding control region (Table 1),which were found in all of the other 30 species involved in the comparative study.The gene arrangements are conserved within Pentatominae.For the 32 mtgenomes of Pentatominae involved in the comparative study,29 are complete with the length ranging from 14 932 (Hoplistoderaincisa) to 16 889 bp (Nezaraviridula).Overall,the A+T content (76.14% on average) of the complete mtgenomes is significantly higher than the G+C content (23.84% on average).The AT-skew values of the whole mtgenome,tRNAs,and control region were greater than 0.Negative GC-skew values were found in the whole mtgenome,the 2nd and 3rd positions of the codons of the PCGs and control region (Fig.1).Conserved gene overlaps and spacers are presented between genes,e.g.,trnW/trnC(-8 bp),atp8/atp6(-7 bp)(expect inDolycorisbaccarum-13 bp),nad4/nad4L(-7 bp),nad4L/trnT(2 bp).The biggest spacer was observed betweentrnS2 andnad1 ranging from 22 bp (Pentatomasemiannulata) to 37 bp (Caystrusobscurus).
3.2 PCGs
Most of the PCGs share the start codon ATN (ATT/ATA/ATG/ATC),and the genescox3 andcytbare all started with ATG in all Pentatominae species (Fig.2).TTG is the common start codon inatp8 andcox1.Only a small number of start codons are GTG (atp8 andatp6) and GTA (nad1).Most of the PCGs are ended with the stop codon TAA/TAG,while the truncated stop codon (T) was also found inatp6,cox1,cox2,cox3,cytb,nad3,nad4,nad5 andnad6.Almost all of the stop codons ofcox2 are ended with a single T residue (expect forStagonomusgibbosus).The 13 PCGs consist of 3 672 codons on average,and the relative synonymous codon usage (RSCU) is shown in Fig.3.Evidently,the most frequently utilized codons are UUA (Leu),UCU (Ser) and CGA (Arg),and they almost show nucleotide bias toward A and T.
The sliding window analysis and evolutionary rate analysis were performed based on the sequences of the 13 aligned PCGs (Fig.4).According to the sliding window analysis,nad2 (the nucleotide diversityPi=0.400),nad6(Pi=0.350),andatp8(Pi=0.343) have apparently higher nucleotide diversity than other genes,whilecox1 has the lowest nucleotide diversity (Pi=0.136)(Fig.4:A).Similar results were observed in further pairwise genetic distance analysis.The genesnad2(D=0.705),nad4(D=0.669),nad6(D=0.538) andatp8(D=0.501) evolve comparatively faster,whilecox1(D=0.155),cox2(D=0.173) andcox3(D=0.177) evolve comparatively slower,in whichDshows the genetic distance (Nei,1972),(Fig.4:B).Average Ka/Ks ratios range from 0.075 forcox1 to 0.820 fornad4,indicating that all PCGs are under purifying selection (Fig.4:B).
3.3 Transfer and ribosomal RNAs
All of the 22 typical tRNA genes range from 52 to 75 bp in length.Most of the tRNA genes can be folded into the typical cloverleaf structure,while some tRNA genes lose their dihydrouridine (DHU) arm and form a loop (i.e.,trnIinAntestiopsisthunbergia,trnS1 andtrnVin all of the Pentatominae species)(Fig.5,takingCarbulacrassiventrisas an example to demonstrate the potential secondary structure of tRNA genes in the mtgenomes of Pentatominae).All tRNA genes use the standard anticodon.The tRNA genestrnT,trnL1,trnL2 andtrnKindicate the lowest variation.In contrast,trnH,trnFandtrnQindicate relatively higher nucleotide substitutions.The two rRNA genes,rrnLandrrnS,were found on the minority strand.The size of the two rRNA genes range from 1 269 to 1 289 bp and 781 to 789 bp,respectively.The average A+T content ofrrnS(77.16%) is smaller than that ofrrnL(78.58%).
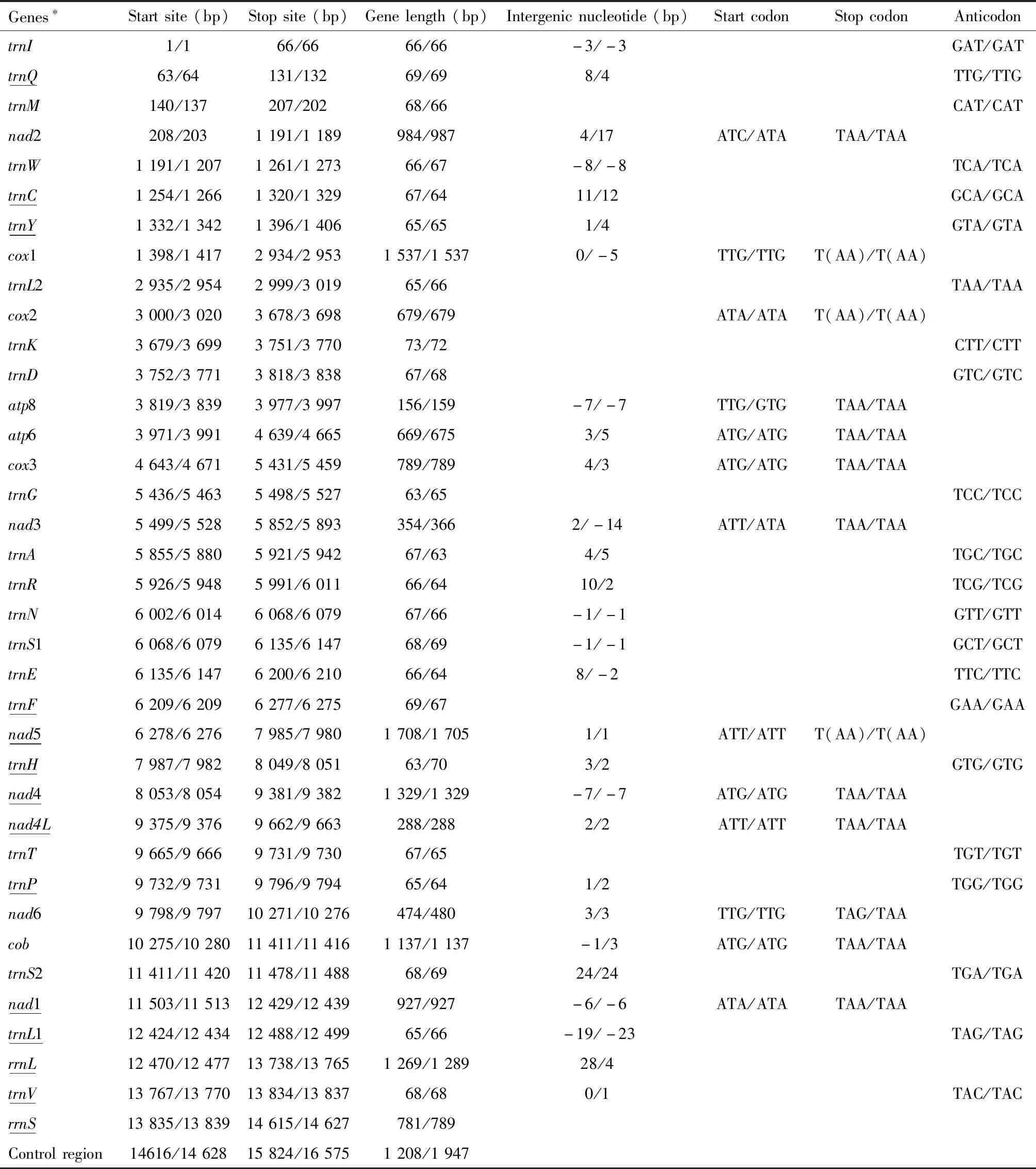
Table 1 Main features of the mitochondrial genomes of Carbula crassiventris and Carpocoris purpureipennis
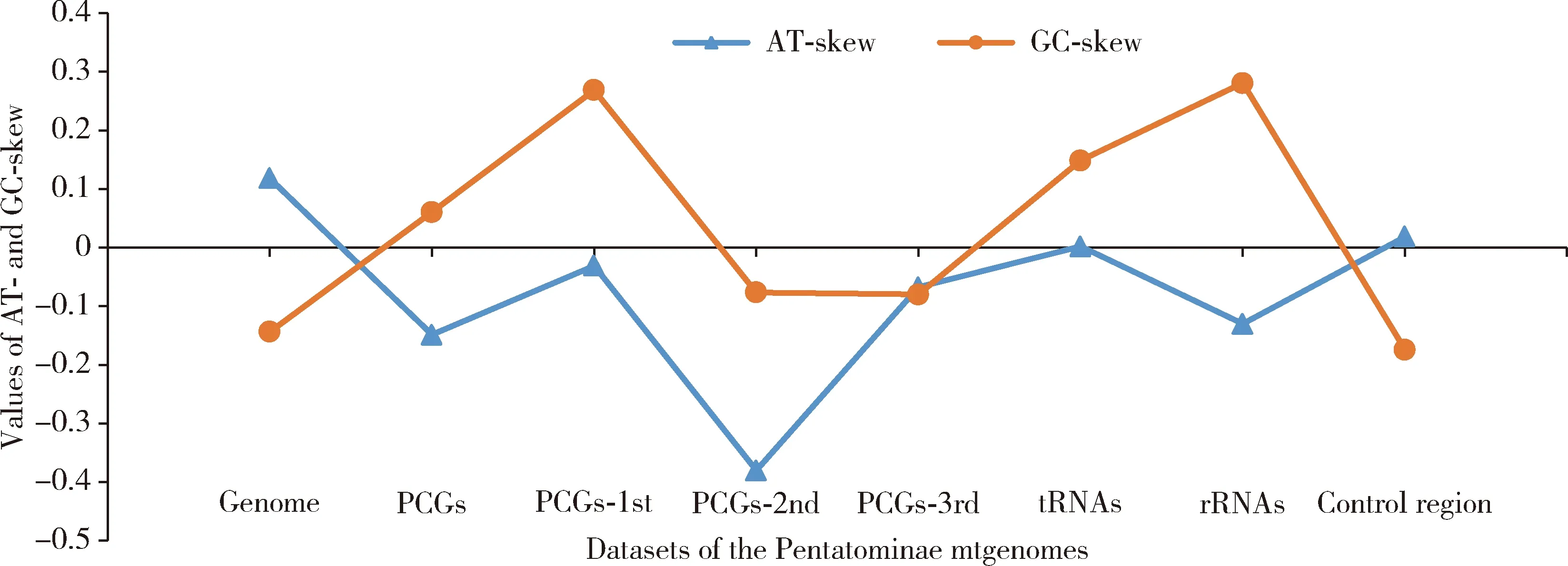
Fig.1 AT-skew and GC-skew of the mitochondrial genomes (mtgenomes) of Pentatominae species PCGs-1st,PCGs-2nd,PCGs-3rd:The 1st,2nd and 3rd positions of the codons of PCGs,respectively.
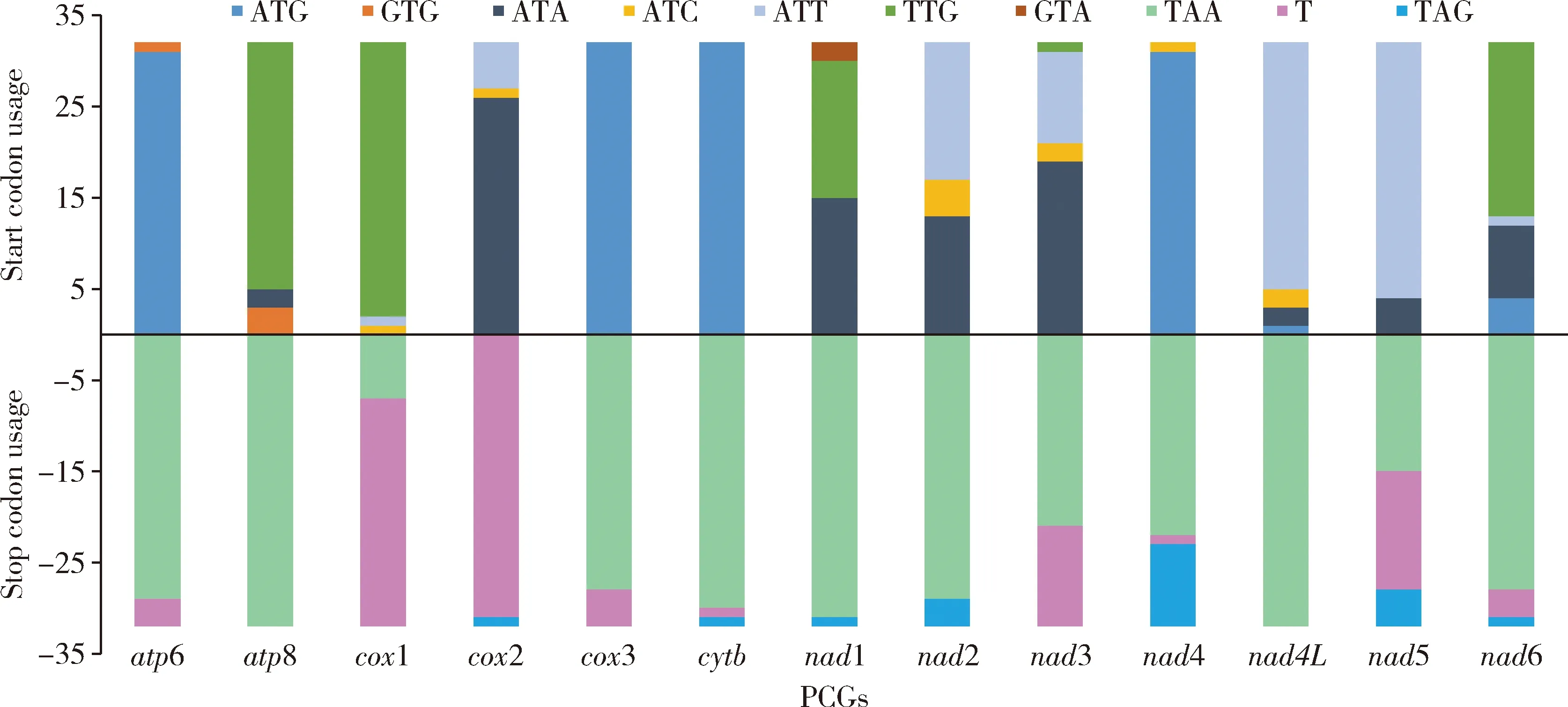
Fig.2 Start and stop codon usage of protein-coding genes (PCGs) in the mitochondrial genomes (mtgenomes) of Pentatominae species

Fig.3 Relative synonymous codon usage (RSCU) of protein-coding genes in the mitochondrial genomes of Pentatominae speciesF:Pro;L1:Leu;L2:Leu;I:Ile;M:Met;V:Val;S2:Ser;P:Pro;T:Thr;A:Ala;Y:Tyr;H:His;Q:Gln;N:Asn;K:Lys;D:Asp;E:Glu;C:Cys;W:Trp;R:Arg;S1:Ser;G:Gly.The stop codon is not given.

Fig.4 Nucleotide diversity (Pi)(A),and genetic distance and nucleotide substitution rates (Ka/Ks) (B) of 13 protein-coding genes (PCGs) in the mitochondrial genomes of Pentatominae speciesKa:Non-synonymous;Ks:Synonymous.
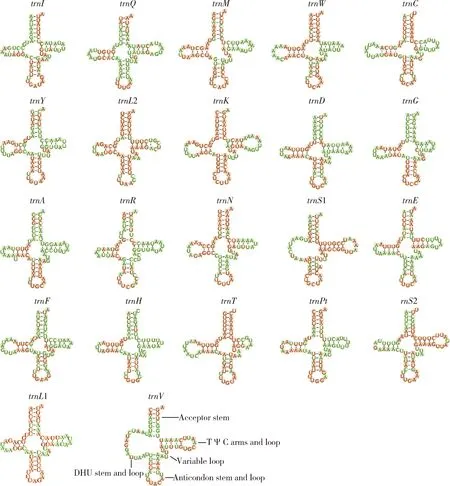
Fig.5 Potential secondary structure of tRNA genes in the mitochondrial genome of Carbula crassiventris The conserved sites within Pentatominae are labeled in red,and the variable sites in green.
3.4 Control region
The size of the control regions varies greatly from 185 (Hoplistoderaincisa) to 2 189 bp (Nezaraviridula),and the A+T content range from 66.58% (Palomenaviridissima) to 82.16% (Erthesinafullo).According to Fig.6 and previous studies (Yuanetal.,2015;Zhaoetal.,2019;Lietal.,2021),most of the mtgenomes have tandem repeats in their control regions,except for four species (Hoplistoderaincisa,Carbulasinica,PalomenaviridissimaandEysarcorisguttiger).Apart fromStagonomusgibbosus,the tandem repeats always appear at the 3′-end of the control region with 113 (inEurydemadominulus) to 788 bp (Catacanthusincarnatus) non-repeat regions located at the 5′-end.In addition,the type,size and copy number of tandem repeats are not fixed among species.

Fig.6 Organization of the control region in the mitochondrial genomes of Pentatominae species that have not been analyzed in previous studies Species without tandem repeats are not shown.The tandem repeats are shown by the blue oval with sequence length and copy number underling.Non-repeat regions are shown by orange box with sequence length inside.
3.5 Phylogenetic relationships
The phylogenetic analyses were performed based on the sequences of the 13 PCGs and 2 rRNA genes.Both the BI and ML methods resulted in the same tree topology:(Menidini+(Hoplistoderini+(Strachiini+((Catacanthini+Pentatomasemiannulata)+(((Sephelini+Halyini)+(Caystrini+(Cappaeini+Placosternumurus)))+((Eysarcorini+Carpocorini)+(Antestiini+Nezarini)))))))(Fig.7).In the phylogenetic analyses,the tribe Eysarcorini and Carpocorini form sister group,but the support value in ML analyses is relatively low (<70).Within each tribe,stable clades are formed with high support values:((Carbula+Eysarcoris)+Stagonomus) and ((Carpocoris+Dolycoris)+Rubiconia),respectively.

Fig.7 Phylogenetic tree of tribes and genera within Pentatominae based on the dataset of sequences of the 1st and 2nd positions of the codons of the 13 protein-coding genes (PCGs) and 2 rRNA genes in the mitochondrial genomes using Bayesian inference (BI) and maximum likelihood (ML) methods
4 DISCUSSION
In this study,we described the mtgenomes ofCarbulacrassiventrisandCarpocorispurpureipennis,and compared them with other 30 species of Pentatominae.The gene arrangements are conserved and consistent with that ofDrosophilayakuba(Clary and Wolstenholme,1985).The sizes of mtgenomes vary greatly from species to species,primarily due to the significant size variation in the control region (Wangetal.,2020;Xuetal.,2021;Yanetal.,2021;Yuanetal.,2021).In our study,the tandem repeats are always located at the 3′-end of the control region within Pentatominae (Fig.6),and this seems relatively conserved within Pentatomoidea (Xuetal.,2021).Although the type,size and copy number of tandem repeats are not fixed among species,we also found some rules:species of Eysarcorini tend to have one type of tandem repeats with length>100 bp;in Carpocorini,two copies of tandem repeats of approximately 50 bp were detected,and this pattern is more similar to that of Nezarini and Antestiini (Yuanetal.,2015;Lietal.,2021).
The most frequently occurring start codon of Pentatominae is ATN,which is similar to that of most Pentatomoidea mtgenomes (Zhaoetal.,2019;Xuetal.,2021).TTG is a common start codon foratp8 andcox1 in Pentatominae (Fig.2),but we also found some differences for the tribes Eysarcorini and Carpocorini.In the tribe Eysarcorini,TTG is the second most frequently used start codon,which is conserved in the genesatp8,cox1 andnad6 (Liuetal.,2019;Lietal.,2021).On the other hand,in the tribe Carpocorini,it is more variable.It is only used forcox1 inDolycorisbaccarum,for two genes (cox1 andnad6) inCarpocorispurpureipennisand for three genes (cox1,atp8 andnad1)Rubiconiaintermedia.
The cloverleaf structure of tRNA genes is most likely conserved across metazoans so as to guarantee the efficiency of transcription (Masta and Boore,2008).For heteropteran mtgenomes,the majority of tRNA genes have a canonical cloverleaf secondary structure,except fortrnS1 andtrnV(Xuetal.,2021).However,truncated tRNAs are not limited to the two tRNA genes described above.InAntestiopsisthunbergia,about 13 nucleotides at the 5′-end of thetrnIare missing,resulting in the lack of the dihydrouridine arm (Zhaoetal.,2021).It was said that mitochondrial tRNA editing plays an important role in recovering the well-paired acceptor stem in metazoans (Lavrovetal.,2000).The posttranscriptional tRNA editing likely exists in heteropteran species and the truncated tRNAs may be functional.
Pentatominae is the most diverse subfamily in the Pentatomidae with its members occurring worldwide.The lack of unique diagnostic characteristics hampers the identification of this subfamily,making it difficult to construct a useful and stable classification.As a result,the classification system of Pentatominae varies from worker to worker (Rideretal.,2018).The scutellum ofCarbulais not as large as that of most eysarcorine genera,and Linnavuori (1982) proposed theCarbulaGroup,includingCarbulaand six other genera based on the characteristics of the carinate mesosternum,ostiolar rugae,and evaporative areas.The taxonomic status ofEysarcorisis also ambiguous,and it was successively placed into four different tribes (Distant,1902;Kirkaldy,1909;Yang,1962;Rider,2006,2018).In the present study,the phylogenetic analyses based on 13 PCGs and 2 rRNA genes strongly support the close relationship betweenCarbula,EysarcorisandStagonomus,which form an independent clade (Fig.7).This result is consistent with the previous chromosomal,morphological,and molecular studies (Zhang and Zheng,2001;Lietal.,2019,2021),and they should belong to the tribe Eysarcorini.Yang (1962) proposed the tribe Dolycorini based on the genusDolycoris.Our study shows thatCarpocorispurpureipennisandRubiconiaintermediaare sisters toDolycorisbaccarum,and this is supported by their current morphological classification and mitochondrial phylogeny (Rideretal.,2018;Liuetal.,2019).It is difficult to separate the Carpocorini from the Eysarcorini and Pentatomini only based on morphological characteristics (Rideretal.,2018).Pentatomini is a large and more poorly defined tribe.The sternal structure ofPlacosternumurusis similar to the pentatomines,but the ostiolar rugae are much shorter or auriculate which could exclude it from Pentatomini.In this study,members of Pentatomini,PlacosternumurusandPentatomasemiannulata,are sister to Cappaeini and Catacanthini,respectively (Fig.7).This indicates thatPlacosternumurusmay not belong to Pentatomini.According to the phylogenetic tree,Carpocorini is clustered to Eysarcorini,but the supporting values are relatively low (Fig.7).Combined with the control region structure,we suggest that further study should be conducted to clarify the relationship of Eysarcorini,Carpocorini,Nezarini and Antestiini.We also observed that the topology of the phylogenetic tree changed slightly compared with the results of our previous study (Lietal.,2021).This may indicate that the relationships among Pentatominae tribes should be reconstructed based on more comprehensive data.
