
(杂)芳烃的C—H硼化研究进展*
2022-11-24 09:46赵琼丽王丽琼
云南化工 2022年11期
张 影,赵琼丽,何 姗,王丽琼
(云南师范大学 化学化工学院,云南 昆明 650500)
近年来,由于C—H功能化可以简化功能分子的合成,因而被广泛研究和使用。有机硼化合物因其在有机合成中具有多功能反应性,对环境、使用者友好,不仅作为高分子、材料、化学传感器以及生物活性分子的重要组成部分,更是在有机合成中起着至关重要的作用[1],它可作为重要的合成中间体,在天然产物、药物和有机材料的构建中起着关键作用,也正因如此,有机硼化合物的高效合成及转化是合成化学、材料化学乃至制药领域中一直是备受关注的研究方向[2]。
由于在合成中使用有机硼试剂能够得到许多益处,因此越来越多的人致力于开发芳香化合物的硼化反应。芳香硼化合物的经典合成方法是利用芳基卤化物进行硼化反应,主要有两类方法,一种是通过卤素金属交换将芳基卤化物转化为有机金属中间体,然后与硼酸酯反应,得到有机硼产物,加酸后,可使有机硼产物水解成硼酸。显然,该途径比碱基敏感、亲电或质子官能团不相容。另一种方法是利用芳基卤化物与硼化试剂的金属催化偶联,即Miyaura硼化[3]。1995年,Miyaura小组[3]首次使用钯催化剂实现了芳基卤化物与双频哪醇二硼烷(B2pin2)的硼酰化。自发现以来,该方案已结合其无数的改进,因其更高的官能团相容性和更温和的反应条件,已被广泛用于制备各种有机硼化合物。然而,在这两种情况下,有机硼化合物的可用性在很大程度上取决于有机卤化物的可用性,当起始材料,特别是高度官能化的有机卤化物难以获得时,便会限制这两种方法的应用。因此,越来越多的研究者致力于开发新的合成路线来构建C—B键,即通过一些其它丰富但相对与硼酰化反应惰性的化学键的裂解而形成C—B键。在过去的几十年中,在研究者们不懈努力下,这一目标获得巨大进展。现如今,许多之前被认为对硼酰化反应是惰性的化学键可直接转换为C—B键,包括C—H、C—O、C—N、C—S、C—F等[4]。
在过去的研究中,C—H活化一直作为研究者比较重视的一方面,因此,在本综述中,将主要对(杂)芳烃构建C—B键的方法进行综述,即催化(杂)芳烃C—H硼酰化反应,其中包括(杂)芳烃的定向与非定向C—H硼酰化。
1 (杂)芳烃非定向C—H硼酰化
对于怎样获得有机硼化物,就原子经济性而言,使用硼酰基团催化取代未活化的C—H键应该是获得有机硼化合物的一种非常有吸引力的方法。这种催化过程最先是由Hartwig组[5]利用铼催化剂Cp*Re(CO)3的光化学活化在烷烃上实现。目前,发现通常由2,2-联吡啶型配体支撑的铱络合物在C—H硼酰化反应中是更优越和实用的催化剂,尤其是对于芳香族原料。值得关注的是,芳烃和杂芳烃的C—H硼酰化是目前使用最广泛的非定向C—H官能化反应,其通常是在没有导向基团的帮助下存在选择性问题。
近年来,人们不断开发新的催化C—H硼酰化体系,发现一些过渡金属配合物是此类转化的有效催化剂。除此之外,无金属催化也可作为此类反应催化剂。因此,以下将以不同催化策略为导向来对其进行详细介绍。
1.1 Ir催化C—H硼化
自从1993年由Marder等首次合成金属铱的复合物以来,铱催化芳基C—H键活化硼化反应得到了迅速发展。1999年,Smith小组[6]第一次报道了以铱合物Cp*(PMe3)Ir(H)(Bpin)(Cp*=五甲基环戊二烯;pin=O2C2H4)催化芳烃C—H硼化。2002年,Smith小组后续研究表明,双齿膦配体,如1,2-双二苯基膦乙烷(dppe)能够显著提高C—H硼酰化的稳定性。此后不久,Ishiyama、Miyaura、Hartwig及其同事报告指出,联吡啶是Ir催化C—H硼酰化的特别有效配体[7]。这些小组通过对铱的前体进行广泛筛选,最终确定了[Ir(cod)OMe]2是迄今为止芳烃C—H硼化最活跃的前体[8]。所对应的催化系统即[Ir(cod)OMe]2、联吡啶配体和四烷氧基二硼化物,此系统也被认为是芳烃C—H硼化的基准。通过这些开创性的研究,以及Hartwig和Sakaki的机理研究,为C—H硼酰化反应领域的发展奠定了坚实基础。
一般来说,芳烃硼酰化的区域选择性很大程度上受空间效应控制,其中硼酰基团结合到最具空间可及性的C—H位置。相反,杂芳烃硼酰化的位点选择性主要由电子效应控制。如在五元环杂环中,会优先在杂原子的α位的C—H键进行硼酰化反应。基于这种反应特性,该催化体系现已用于制备各种官能化(杂)芳基硼酸衍生物,并可对该衍生物进行进一步衍生。
因此,(杂)芳烃的C—H硼酰化可以说提供了一个灵活可靠的平台,通过获得芳基硼酸酯,继而实现了许多间接的芳香族C—H键功能化反应。如在2010年,Hartwig小组[9]首次公开了铜介导的芳基硼酸和芳基硼酸酯的氧化氰化反应。然后,在Ir催化的C—H硼酰化之后,采用这种氰化方法,可以将简单的1,3-二取代芳烃一锅合成相应的间取代芳腈(见图1)。此类反应,可使用于各种官能团,如卤化物、酮、酯、酰胺和保护醇等,都能与之兼容。根据这种方法,可直接由2,6-二甲基苯酚合成艾滋病药物依特拉维林的中间体4-氰基-2,6-二甲基苯酚,产率可达58%。同年,Marder、Steel小组[10]发表了一种利用微波辅助,一锅串联Ir催化的C—H硼酰化/Rh催化的烯酮序列加成反应,通过溶剂选择选择性地提供芳基取代的酮或相应的醇。在此过程中,当使用不可氧化溶剂如甲基叔丁基醚(MTBE)作为溶剂时,得到酮产物。相反,如果将反应溶剂改为异丙醇(IPA),则可实现酮的进一步还原反应,并选择性形成醇产物。

图1 通过Ir催化的硼酰化反应实现芳烃的氰化反应
1.2 Rh催化C—H硼化
虽然Rh催化的(杂)芳烃的C—H硼酰化几乎与Ir催化的对应物同时被发现。然而在过去几十年中,对Rh催化的C—H硼酰化反应的进一步探索和应用却远远不及其他。虽说对Rh催化的研究没有对Ir等多,但仍不断有研究者对以铑络合物为催化剂的催化体系进行探索,如在2015年,Beller小组[11]公开了一种以反式[Rh(PMe3)2(CO)Cl]为活性光催化剂,HBpin为硼源的(杂)芳烃C—H硼酰化的光催化合成路线。指出在温和的条件下(r.t.-40 ℃),可以通过该途径获得多种含吸电子和给电子取代基的硼酰化(杂)芳基。
1.3 Ni催化C—H硼化
2015年,Chatani小组[12]和Itami小组[13]几乎同时发表了他们在镍催化的(杂)芳烃C—H硼酰化方面的研究成果。他们两组通过调整催化剂前体、配体、碱、溶剂和硼酰化试剂的组合实现催化硼酰化。Itami小组提出使用镍催化芳香族C—H硼酰化反应的主要内容为:即在[Ni(cod)2]催化下,CsF作为添加剂,以PCyp3为配体,B2pin2为硼酰化试剂,可将苯和吲哚衍生物进行硼酰化(见图2)。而Chatani小组所开发的反应体系指出N-杂环卡宾配体对于有效转化至关重要,N-环己基取代衍生物是最佳配体。在两者的研究中均反映出,当在这些硼酰化条件下使用苯基底物时,需要大量过量的芳烃才能实现高效率,所取代芳烃的区域选择性基本上由空间效应控制,主要提供间位化和对位化产物。当使用吲哚时,以吲哚为限制试剂,硼酰化反应效率更高,且硼酰基区域主要是吲哚的C2位。
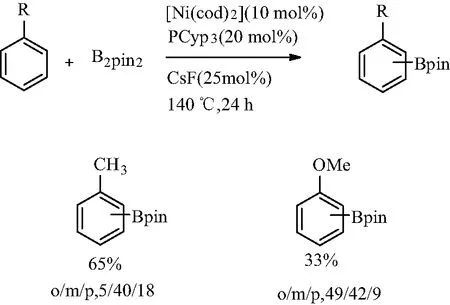
图2 镍催化C—H硼化反应
1.4 无金属催化C—H硼化
在过去几十年中,过渡金属催化剂,特别是贵过渡金属催化剂在芳香族C—H硼酰化反应中发挥了重要作用。尽管过渡金属催化在这些过程中表现出很高的效率,但无金属芳香族C—H硼酰化出现也显示出了它的独特魅力。如在2020年,张华小组[14]提出了以一种新型的无金属C—H硼酰化,主要内容是使用简单廉价的BF3·Et2O作为催化剂,稳定的B2pin2作为硼源,吲哚、多种取代吲哚或其他杂环芳烃作为合适的底物,此方法能够以良好产率获得相应的C2硼酰化产物(见图3)。

图3 无金属催化芳香族C—H硼化反应
2 (杂)芳烃定向C—H硼酰化
上文已指出,催化芳烃硼酰化反应的大部分区域选择性主要由空间因素控制,然而,由于预期的官能化有机硼烷具有巨大的合成潜力,因此现在许多研究小组专注于开发定向位点选择性硼酰化反应。接下来我将对此方面进行介绍。
2.1 Pd催化(杂)芳烃定向C—H硼酰化
钯基催化剂尚未广泛用于C—H硼酰化反应,这是由于所得产物(即硼酯)易于通过与Pd(II)物种的转化分解。然而,在2012年,Fu等[15]人提出了钯催化的定向邻硼酰化的实例,即以Pd(OAc)2为催化剂,苯醌为氧化剂,不需要惰性气氛,便可在温和条件下实现乙酰苯胺邻硼酰化的完全区域选择性,并获得良好的产率[15]。由于形成了内部CO—B键,硼片段采用四面体配位,这使得乙酰基不能用于进一步定向的邻硼酰化(见图4)。
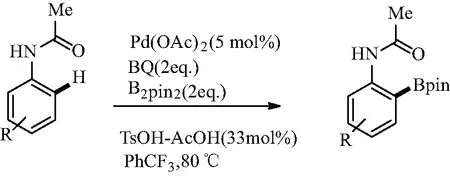
图4 钯催化乙酰苯胺在酸性条件下的单选择性C—H硼酰化
2.2 Rh催化(杂)芳烃定向C—H硼酰化
2014年,Braun[16]发表了在温度为40℃时,以高活性硼酰铑(I)络合物[Rh(Bpin)(PEt3)3]催化含SCF3芳烃C—H硼酰化反应,硼酰基可定向选择于与SCF3基团相连C的邻位,故该组认为基团SCF3可能起到导向基团的作用,且其具有高区域选择性(见图5)。而在室温下用等摩尔量的[Rh(Bpin)(PEt3)3]处理苯基三氟甲基硫化物时,反应产生邻硼酰化和邻二硼酰化产物,以及络合物[Rh(H)(PEt3)]。该小组认为发生了二次催化硼酰化反应的原因是由于,通过[Rh(H)(PEt3)]和硼酰化试剂的相互作用再生出了[Rh(Bpin)(PEt3)3]。
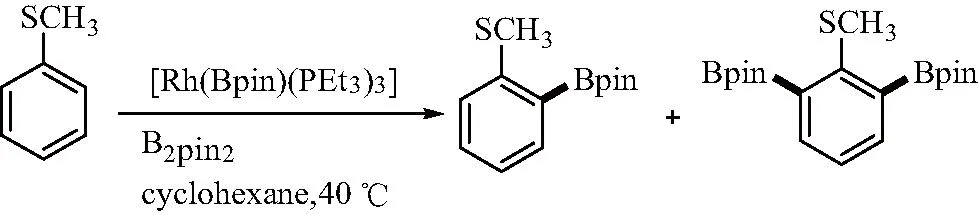
图5 Rh催化含SCF3芳烃的C—H硼酰化反应
3 结论与展望
有机硼化合物的合成作为近几十年来有机合成中的研究热点,随着科学家们对有机硼化合物的合成研究不断深入,使得有机硼化合物的合成反应不断取得突破,新的合成方法不断被开发出来。然而,目前C—H键硼化仍然存在一些限制,如(1)多数反应体系得到单双取代混合物;(2)反应底物类型有限;(3)与铱基催化剂相比,使用非贵催化剂的已知方法对未活化芳烃的能力仍然较差等。因此,相信在不久的将来,(杂)芳基硼酸类化合物的合成方法将会取得更大的突破,并能够在有机合成、药物化学及材料科学中拥有更广泛的应用。
猜你喜欢
食管疾病(2022年1期)2022-11-26
中南民族大学学报(自然科学版)(2022年6期)2022-11-02
分子催化(2022年1期)2022-11-02
科学家(2021年24期)2021-04-25
中南民族大学学报(自然科学版)(2020年6期)2020-12-22
天然产物研究与开发(2019年8期)2019-09-05
中国塑料(2015年10期)2015-10-14
橡胶工业(2015年9期)2015-08-29
橡胶工业(2015年6期)2015-07-29
橡胶工业(2015年4期)2015-07-29
