
UPLC-MS/MS 法测定白酒中三氯蔗糖含量的不确定度分析
2022-11-24 10:06:14黄永桥吴新文张世芹简银池毛敏霞
酿酒科技 2022年11期
黄永桥,吴新文,张世芹,简银池,毛敏霞
(贵州省检测技术研究应用中心,贵州贵阳 550014)
三氯蔗糖(Sucralose)又名蔗糖素,是一种非营养型强力甜味剂,为蔗糖甜度的600 倍,无能量,热量值为零,为减肥人群及血糖异常人群所喜爱,被广泛应用于饮料、调味料、糕点等食品中[1-3]。研究表明,三氯蔗糖可以影响人体代谢,具有一定的慢性毒性、生殖毒性,会增加患癌症的风险,并且对生态环境存在潜在影响[4-6]。GB 2760—2014《食品安全国家标准食品添加剂使用标准》中规定配制酒中三氯蔗糖的最大允许限量为0.25 g/kg,但在白酒中不得添加。
目前三氯蔗糖的检测方法有:液相色谱法[7-8]、液相色谱-串联质谱法[9-10]、气相色谱法[11]、滴定法[12]和离子色谱法[13-14]等。液相色谱-串联质谱法具有较强的选择性和抗干扰能力、较高的灵敏度等优点。现有三氯蔗糖的检测方法为GB 22255—2014《食品安全国家标准食品中三氯蔗糖(蔗糖素)的测定》,现阶段采用液质联用法测定白酒中三氯蔗糖的不确定度评定的报道较少。研究参照现有测量分析过程中不确定度评定方法和要求,对超高效液相色谱-串联质谱法测定白酒中三氯蔗糖含量的不确定度进行分析评定[15-16]。根据分析测量过程中不确定度的来源,建立的数学模型并对其进行不确定度评估,计算合成相对不确定度,得到最终的扩展不确定度,以期为测量结果的准确性提供科学依据。
1 材料与方法
1.1 材料、试剂及仪器
样品:市售蒸馏白酒样品。
试剂及耗材:乙腈(色谱纯),德国Merck 公司;甲酸(色谱纯),上海安普实验科技股份有限公司;三氯蔗糖(纯度>99%),上海安谱实验科技股份有限公司。
仪器设备:Agilent 1290 超高效液相色谱仪、Agilent 6470 QQQ 三重串联四级杆质谱(配有ESI源),美国Agilent 公司;LT2002 电子天平,常熟市天量仪器有限公司;UMV-2 多管涡旋混合器,北京普立泰科仪器有限公司;Milli-Q 超纯水机,美国Millipore 公司;0.22 μm 有机滤膜,上海安谱实验科技股份有限公司。
1.2 试验方法
1.2.1 标准溶液配制
标准储备溶液配制:准确称取三氯蔗糖标准品10 mg(精确至0.0001 g),纯水溶解并定容至10 mL,得到浓度为1.0 mg/mL的标准储备溶液。
1.2.2 仪器条件
1.2.2.1 色谱条件
ZORBAX Eclipse Plus-C18(50 mm×2.1 mm,1.8 μm,美国Agilent 公司)色谱柱;流动相A:0.1%甲酸水溶液,B:乙腈,流速:0.3 mL/min,柱温:40 ℃,进样体积:2.0 μL,梯度洗脱程序:0.0~0.5 min,20 %B;0.5~1.0 min,20 %~80 %B;1.0~2.0 min,80 %B;2.0~2.5 min,80 %~20 %B;2.5~3.5 min,20%B。
1.2.2.2 质谱条件
电喷雾离子源,负离子扫描;多反应监测;毛细管电压:4.5 kV;雾化器压力:45 psi;干燥气流速:7 L/min;干燥气温度:325 ℃;鞘气流速:10 L/min;鞘气温度:300 ℃;三氯蔗糖质谱参数:锥孔电压130 V,母离子m/z 397,定量离子m/z 360.8,碰撞能量8 eV,母离子m/z 395,定量离子m/z 358.8,碰撞能量6 eV。
1.2.3 样品前处理
准确称取5 g 试样(精确至0.01 g)置于50 mL烧杯中,沸水浴上蒸干,残渣用11%乙腈溶液溶解并转移至25 mL 容量瓶中,定容后过膜,供液相色谱-串联质谱分析。
1.2.4 建立不确定度数学模型

式中:X——试样中三氯蔗糖的含量,μg/kg;
C——三氯蔗糖质量浓度,ng/mL;
V——试样溶液定容体积,mL;
m——试样质量,g;
1.2.5 测量不确定度来源
测量三氯蔗糖的不确定度来源主要有标准溶液引入的不确定度和样品前处理引入的不确定度,标准溶液引入的不确定度包括:标准物质纯度、标准储备液、标准中间液和标准工作液的配制、标准曲线拟合;样品前处理引入的不确定度包括:样品称样、样品前处理、测量重复性和加标回收率。
注:文中后续阳性样品表示经测定发现白酒样品中含有三氯蔗糖,阴性样品表示经检验未发现白酒样品中含有三氯蔗糖。
2 结果与分析
2.1 标准溶液配制过程引入的不确定度urel(S)
2.1.1 标准物质引入的不确定度
根据标准物质证书上的信息,三氯蔗糖标准物质纯度为98.79 %,不确定度为±1 %,符合均匀分布,包含因子k=,因此相对标准不确定度为:
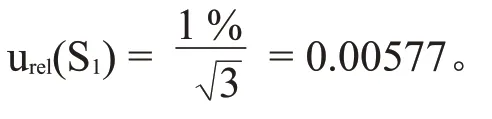
2.1.2 标准储备液配制引入的不确定度urel(S2)
2.1.2.1 标准品称量引入的不确定度urel(w)
称量所用分析天平检定证书给出允差为±0.1 mg,符合均匀分布,k=,当标准品称量为10 mg,引入相对标准不确定度为:

2.1.2.2 标准储备液配制过程中器具引入的不确定度urel(std1)
标准储备液配制过程引入不确定度有10 mL单标线容量瓶(A 级)的容量允差和实验室温度变化引起的溶剂和玻璃容器体积膨胀。根据JJG 196—2006《常用玻璃量器检定规程》中可知,10 mL 单标线容量瓶(A 级)容量允差为±0.020 mL,实验室温度在(20±5)℃之间变动时,查得水和玻璃的膨胀系数分别为0.000208/℃和0.0000250/℃,玻璃的体积膨胀系数相较水的体积膨胀系数可忽略不计,符合均匀分布k=,则10 mL 单标线容量瓶(A 级)的容量允差不确定度urel(n1)和水的体积膨胀不确定度urel(n2)为:
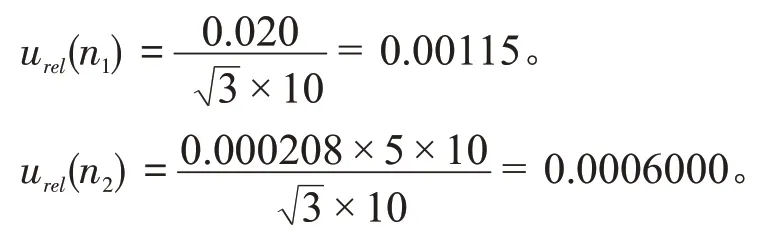
因此,标准储备液配制过程中器具引入的相对标准不确定度为:

综上,标准储备液配制引入的相对标准不确定度为:

2.1.3 标准中间液配制引入的不确定度urel(S3)
吸取标准储备溶液0.10 mL 于100 mL 容量瓶中,用水稀释并定容至刻度,得到1.0 μg/mL标准中间溶液。
配制过程引入不确定度有1 mL分度吸量管(A级)的容量允差、10 mL 单标线容量瓶(A 级)的容量允差和因实验室温度变化引起的溶剂体积膨胀。根据JJG 196—2006《常用玻璃量器检定规程》中可知,1 mL 分度吸量管(A 级)容量允差和10 mL单标线容量瓶(A 级)容量允差为±0.008 mL 和±0.020 mL,实验室温度在(20±5)℃之间变动时,水的体积膨胀系数为0.000208/℃,符合均匀分布k=,标准中间液配制过程引入的不确定度见表1。
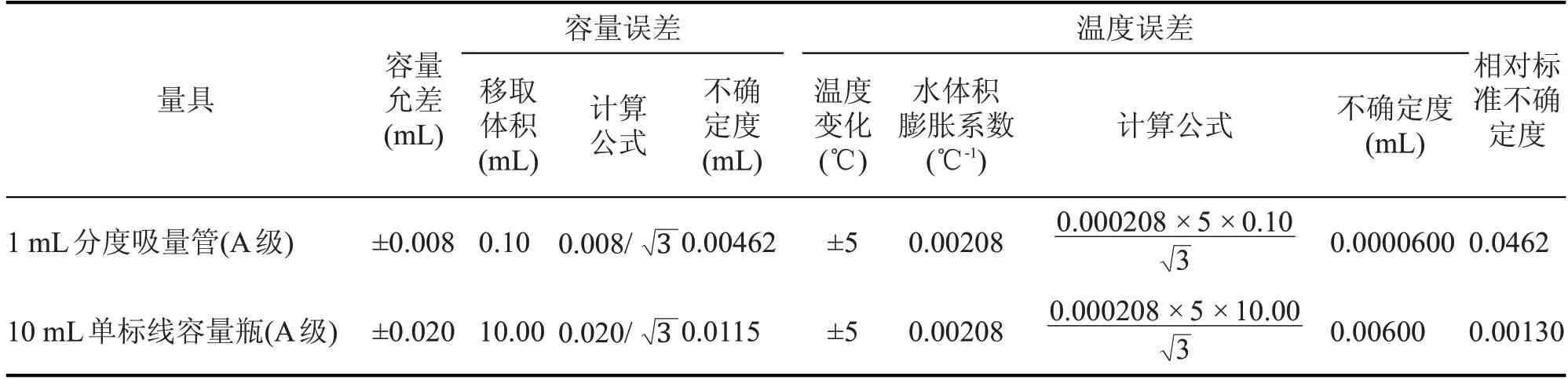
表1 标准中间液配制过程引入的不确定度
因此,标准中间液配制引入的相对标准不确定度为:

2.1.4 标准工作液配制引入的不确定度urel(S4)
分别吸取标准中间液5 μL、10 μL、20 μL、50 μL、100 μL、200 μL 于10 mL 容量瓶中,用水稀释并定容至刻度,混匀,得到质量浓度为0.5 ng/mL、1 ng/mL、2 ng/mL、5 ng/mL、10 ng/mL、20 ng/mL 的标准系列工作液。
配制过程引入不确定度有不同量程移液器的容量允差、10 mL 单标线容量瓶(A 级)的容量允差和因实验室温度变化引起的溶剂体积膨胀。根据JJG 646—2006《移液器检定规程》、JJG 196—2006《常用玻璃量器检定规程》和实验室温度在(20±5)℃之间变动时水的体积膨胀系数,符合均匀分布k=,标准工作液配制过程引入的不确定度见表2。
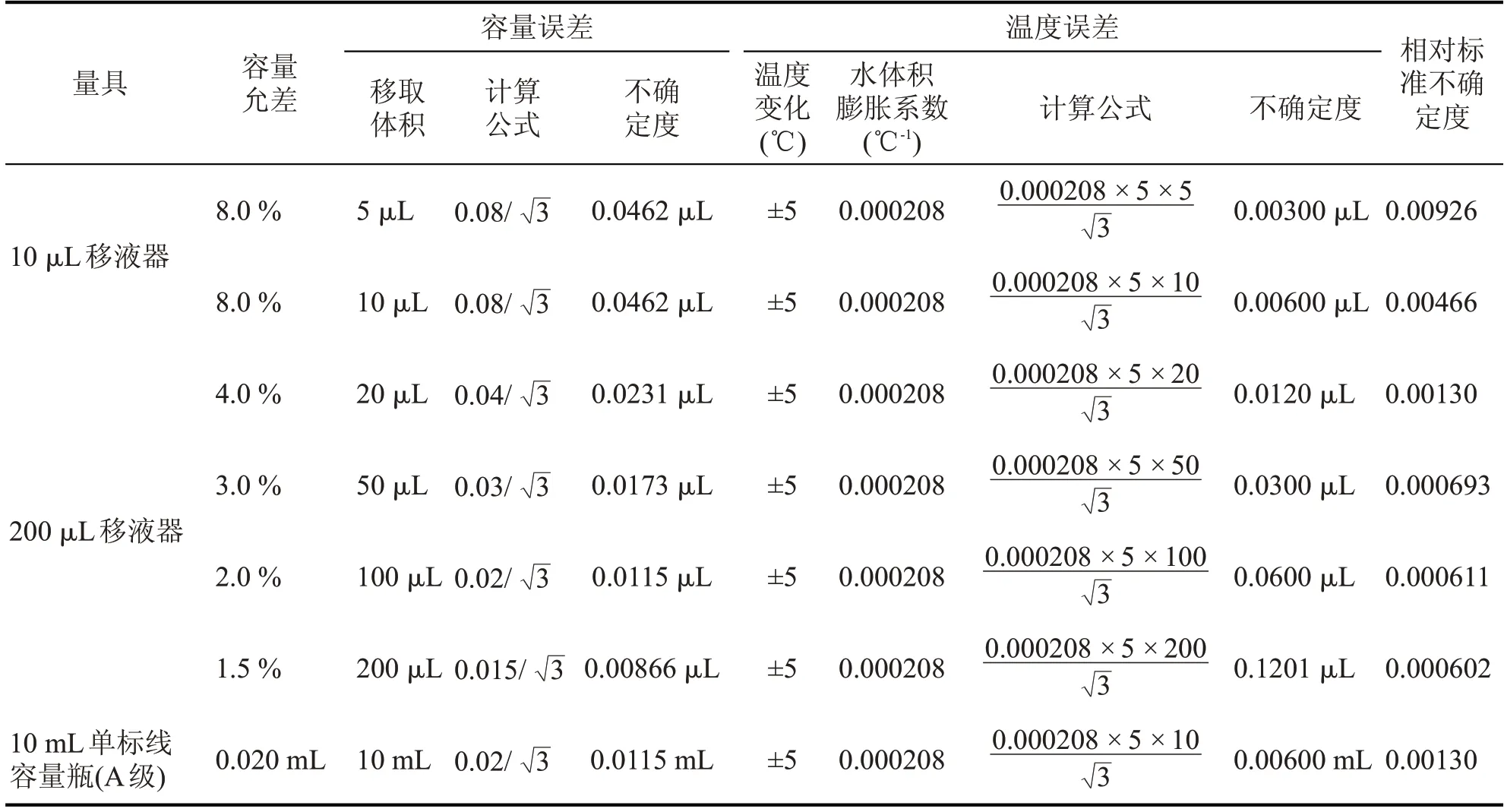
表2 标准工作液配制过程引入的不确定度
因此,标准工作液配制引入的相对标准不确定度为:

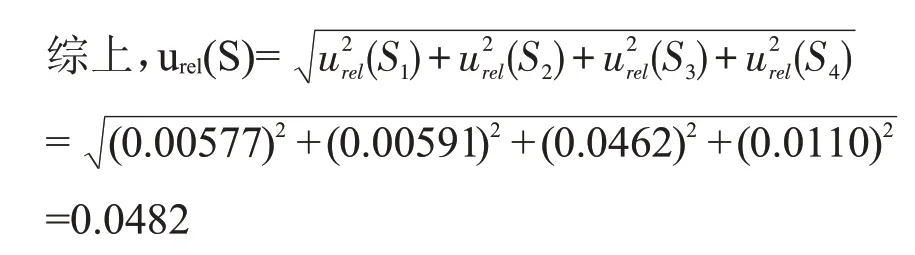
2.2 标准曲线拟合引入的不确定度urel(C)
配制的标准工作液经液相色谱-串联质谱仪进行测定,重复测定3 次,以质量浓度对其峰面积通过直线拟合得到标准曲线,线性回归方程:y=159478c-111279,相关系数0.998。测定结果见表3。

表3 标准工作液峰面积测定结果
选取阳性样品重复测定6 次,将峰面积带入线性方程校正其含量,测得阳性样品中三氯蔗糖平均含量为C0=12.18 ng/mL,标准曲线拟合引入的标准不确定度计算按公式(2),相对标准不确定度计算按公式(3)。
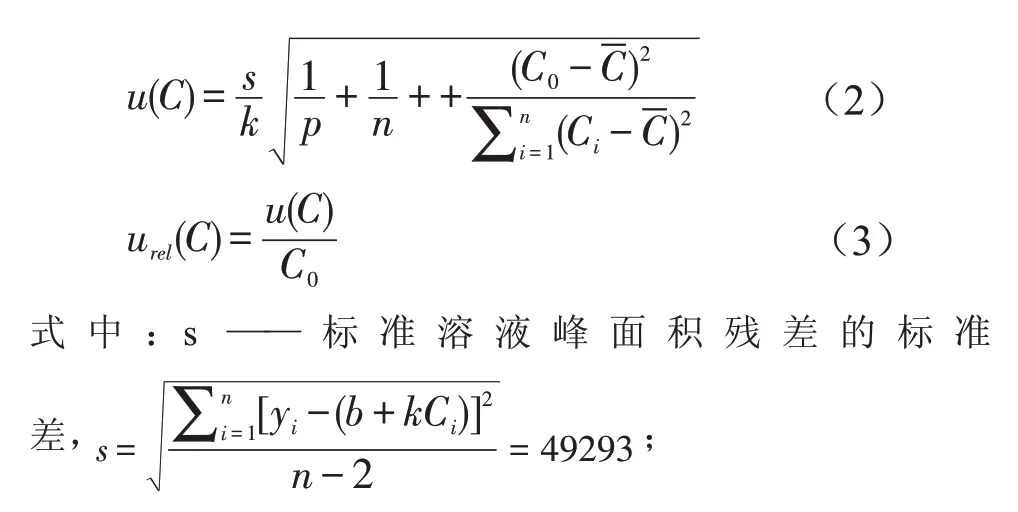
k——线性方程斜率,k= 159478;
b——线性方程截距,b=- 111279;
n——样品平行测定次数,n=6;
p——标准工作液各浓度点重复测定的次数,每个浓度点重复测定3次,p=18;C0——样品溶液的平均质量浓度,C0=12.18 ng/mL;

经计算,样品测定引入的标准不确定度u(C)为0.180 ng/mL,相对标准不确定度urel(C)为0.0148。
2.3 样品前处理引入的不确定度urel(P)
2.3.1 称样引入的不确定度urel(P1)
称取5 g 白酒试样(精确至0.001 g),根据JJG 1036—2008《电子天平检定规程》,最大允许误差为±0.005 g,符合矩形分布k=,则相对标准不确定度为:

2.3.2 样品定容引入的不确定度urel(P2)
样品最终使用11%乙腈溶液溶定容至25 mL,根据JJG 196—2006《常用玻璃量器检定规程》,25 mL 单标线容量瓶(A 级)容量允差为±0.030 mL,20 ℃时乙腈和水的体积膨胀系数分别为0.00137/℃和0.000208/℃,用于定容的11 %乙腈溶液溶的体积膨胀系数为0.000336/℃,服从矩形分布k=,则25 mL 单标线容量瓶(A 级)的容量允差不确定度urel(n3)和11 %乙腈溶液溶的体积膨胀不确定度urel(n4)为:
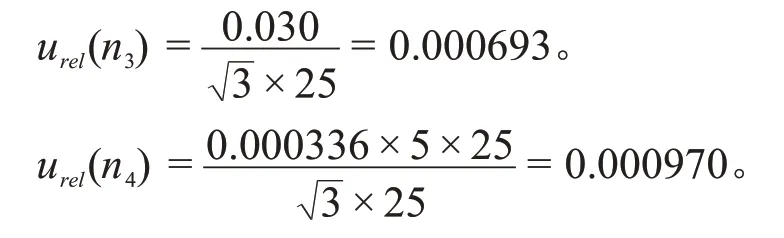
因此,样品定容过程引入的相对标准不确定度为:
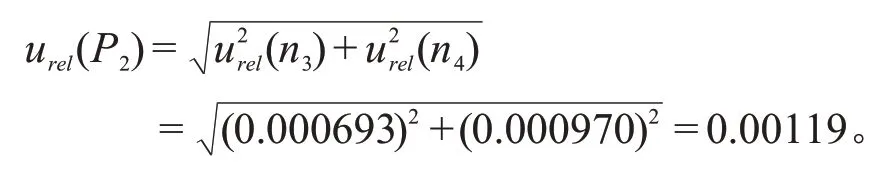
综上,样品前处理引入的不确定度为:

2.4 测量重复性引入的不确定度urel(A)
同时称取阳性样品6份,按照1.2前处理方法进行前处理,计算6 次重复性结果和平均值。结果分别为59.5 μg/kg、59.9 μg/kg、59.7 μg/kg、61.9 μg/kg、60.0 μg/kg、62.4 μg/kg,平均值为60.6 μg/kg,相对标准不确定度计算按公式(4)。

经计算,测量重复性引入的相对标准不确定度urel(A)为0.00829。
2.5 回收率引入的不确定度urel(R)
取阴性样品进行加标回收试验,按1.2 样品前处理进行处理,平行测定6 次,加标回收率统计结果见表4。
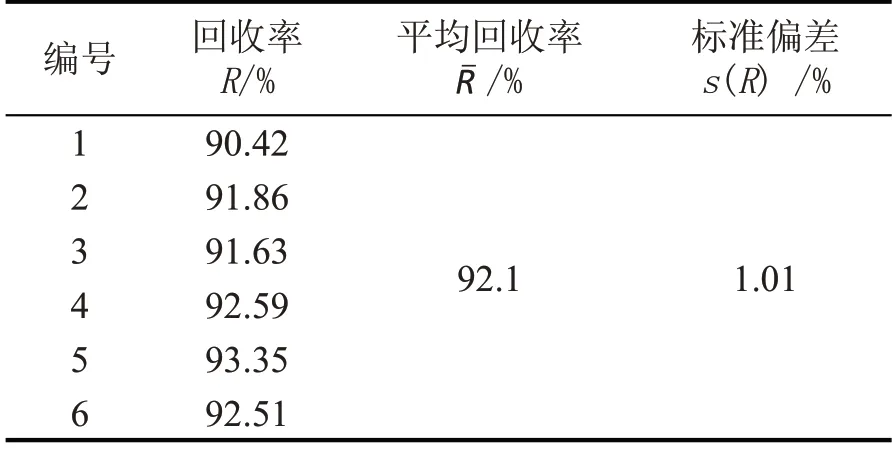
表4 加标回收试验结果(n=6)
加标回收率引入的相对标准不确定度为:
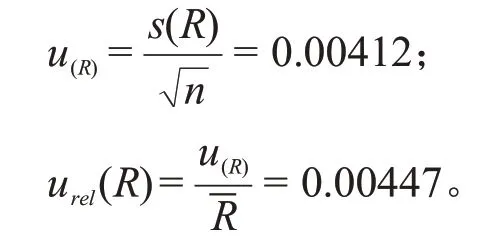
为确定在三氯蔗糖含量的计算中是否需要折算平均回收率,根据CNAS—GL006《化学分析中不确定度的评估指南》,用t检验来确定平均回收率与1.0是否有显著性差异:

查JJF 1059.1—2012《测量不确定度评定与表示》t 分布表,取95 %置信度,n-1 自由度的临界值t95(5)为2.57,故t>t95(5),说明平均回收率与1.0 具有显著性差异,因此在结果计算中需采用回收率对结果进行修正。
2.6 分析过程中不确定度分量及合成相对标准不确定度urel(X)
经过对白酒中三氯蔗糖分析过程中不确定度进行分析和计算,过程中各相对标准不确定度来源、类型和数值见表5。
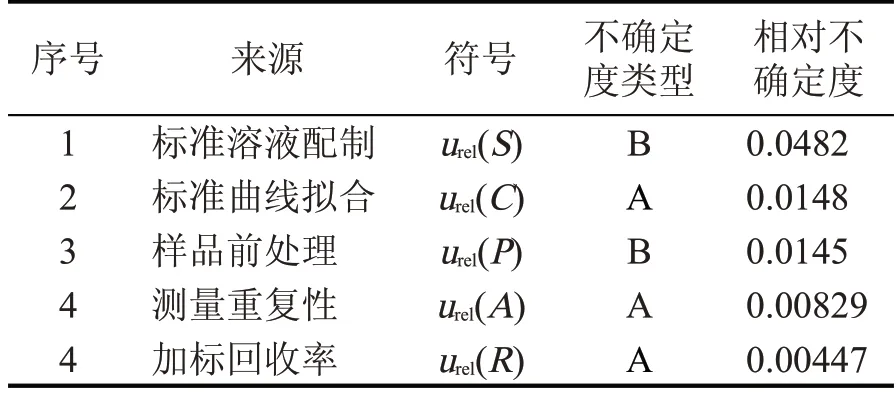
表5 不确定度分量
白酒中三氯蔗糖分析过程中的合成相对标准不确定度为:
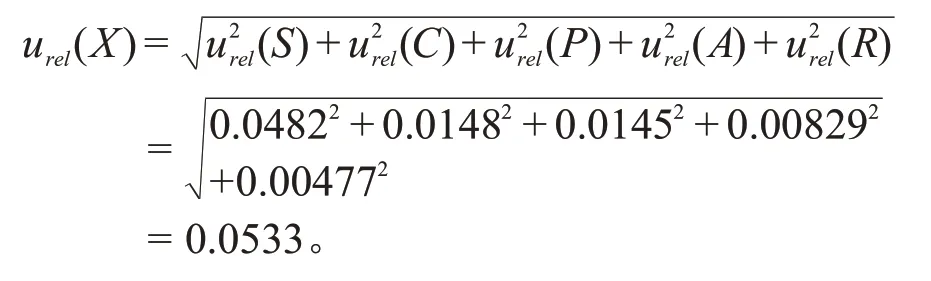
2.7 扩展不确定度
依据JJF 1135—2005《化学分析测量不确定度评定》,取95%置信水平,扩展因子k=2,白酒中三氯蔗糖含量的平均值为60.6 μg/kg,则扩展不确定度:
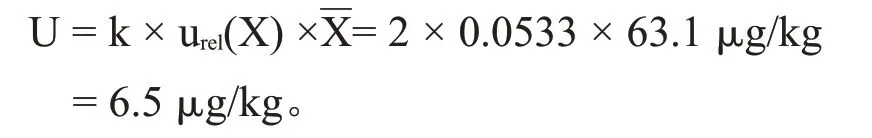
2.8 测量结果及不确定度报告
超高效液相色谱-串联质谱法测定白酒中三氯蔗糖含量,根据不确定度评定结果,当称样量为5 g,置信度为95 %时k=2,测定结果为:(60.6±6.5)μg/kg,k=2。
3 结论
采用UPLC-MS/MS 法测定白酒中三氯蔗糖含量,通过对测定过程中不确定度来源进行分析并量化,结果表明,影响白酒中三氯蔗糖测定结果不确定度的因素主要有:标准溶液配制、标准曲线拟合、样品前处理、测量重复性和加标回收率,其中测量重复性和加标回收率的不确定度对合成相对标准不确定度的贡献较小,标准溶液配制和标准曲线拟合不确定度评定结果最大。因此,在测定白酒中三氯蔗糖时应注意操作的规范性,选择适当量程的量具,准确配制标准溶液,定容准确,减少测量过程不确定度,提高检测结果的准确度和可信度。
猜你喜欢
当代水产(2021年10期)2022-01-12 06:20:40
制造技术与机床(2017年9期)2017-11-27 02:14:23
食品与机械(2017年5期)2017-07-05 13:24:36
环境科技(2016年4期)2016-11-08 12:18:58
电源技术(2015年7期)2015-08-22 08:48:52
西南石油大学学报(自然科学版)(2015年3期)2015-04-16 05:12:08
电测与仪表(2015年7期)2015-04-09 11:40:30
电测与仪表(2014年9期)2014-04-15 00:27:16
电测与仪表(2014年11期)2014-04-04 09:21:40
河南城建学院学报(2014年4期)2014-02-27 07:08:52
