
Fe掺杂对Ni基催化剂催化甲烷干重整的影响
2022-11-23 08:16潘伟滔
煤气与热力 2022年11期
张 玉, 潘伟滔, 周 阳
(佛燃能源集团股份有限公司, 广东 佛山 528200)
1 概述
氢气因质量热值高、能量密度高、燃烧产物无污染、生产原料容易获得等特点,已成为一种很有发展前景的新兴能源[1]。未来几十年,氢燃料电池将广泛应用于交通运输和便携式、固定式电源[2]。
天然气资源丰富,甲烷是天然气的主要成分,氢元素的质量占比高,通常是制备氢气的首选原料。目前,工业上甲烷制氢的主要方法有甲烷重整、部分氧化以及裂解,如甲烷水蒸气重整、甲烷自热重整、甲烷干重整和甲烷直接催化裂解等[3-4]。甲烷直接催化裂解制备H2是一种很有前途的制氢方法,它不仅可以产生不含CO的氢气,而且可以储存生成的C供其他用途。然而,与其他制氢方法相比,甲烷直接催化裂解制氢的能耗和成本较高,很难大规模应用到工业领域。因此,甲烷重整是利用甲烷制备氢气的较好选择。在众多重整反应中,甲烷干重整(DRM)在近年来受到持续的关注,这是因为DRM有着较低的操作成本,而且它还可以把两种温室气体CH4和CO2转化成合成气,缓解了温室效应[5-7]。不仅如此,DRM生成的合成气H2与CO的物质的量比为1∶1,是一种理想的费托合成或含氧衍生物合成的原料[8]。目前DRM反应中普遍使用镍基催化剂,实验研究表明,向镍基催化剂中掺入Fe后,Ni和Fe会形成Ni3Fe,Ni3Fe催化剂的催化性能明显优于纯镍基催化剂[9]。为了进一步了解Ni3Fe催化剂催化性能提高的原因,本论文通过密度泛函理论,研究在Ni3Fe(111)和Ni(111)表面上甲烷脱氢过程,确定各步骤反应物和生成物的稳定构型,并计算不同温度下各反应的速率常数,比较Ni3Fe(111)和Ni(111)表面甲烷脱氢反应的难易程度。
2 计算模型设置与参数定义式
2.1 计算模型设置
Ni在室温下能相对稳定存在的晶胞模型通常属于面心立方晶胞,而Ni(111)晶面对于催化反应来讲是最稳定的表面[10],因此后续计算选用Ni(111)晶面作为Ni金属表面模型。在搭建表面模型时选用4层金属原子构成平板结构,采用了被广泛使用的p(2×2)结构[11-12]。为了满足密度泛函理论(DFT)计算所要求的周期性边界条件,用15×10-10m厚的垂直于表面的真空层将各层原子隔开,以确保层与层之间的相互作用足够小。表面形态学的研究表明,Ni(111)表面上有4个高对称吸附位:顶位(Top site,简称Top)、桥位(Bridge site,简称Bridge)、六角密堆积洞位(Hexagonal close packed site,简称Hcp)、面心立方洞位(Face-centered cubic site,简称Fcc)。Ni(111)表面模型和4个高对称吸附位见图1,蓝色球代表第1层Ni原子,绿色球代表第2层Ni原子,红色球代表第3层Ni原子。
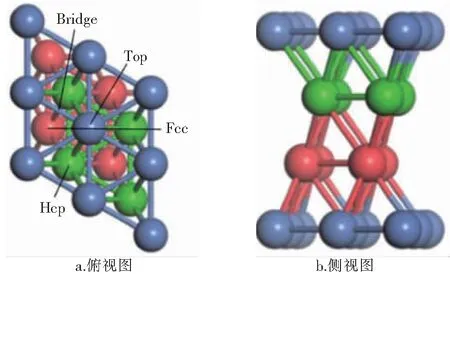
图1 Ni(111)表面模型和4个高对称吸附位
实验和理论证明,当Fe的摩尔分数小于60%时,Ni-Fe合金具有面心立方结构[13-14]。因此,Ni3Fe双金属合金采用L12 Fcc型结构,在Ni晶胞模型的基础上用Fe原子替换Ni原子,其中Ni原子位于面心位置,Fe原子占据单元格的角落。得到的Ni3Fe(111)表面模型有8个高对称吸附位,分别记为Top Fe、Top Ni、Bridge Ni2、Bridge NiFe、Hcp Ni3、Hcp Ni2Fe、Fcc Ni3、Fcc Ni2Fe。Ni3Fe(111)表面模型和8个高对称吸附位见图2,蓝色球代表第1层Ni原子,绿色球代表第2层Ni原子,红色球代表第3层Ni原子。紫色球代表第1层Fe原子,黄色球代表第2层Fe原子,粉色球代表第3层Fe原子。

图2 Ni3Fe(111)表面模型和8个高对称吸附位
本文大部分的计算都是采用CASTEP软件包完成,过渡态的相关计算使用DMol3软件包。采用广义梯度近似密度泛函理论探究甲烷在纯Ni和Ni-Fe双金属表面的吸附解离行为,通过Perdew-Bruke-Ernzerhof(PBE)泛函校正电子与电子相关作用的交换相关能。为了使得整个体系的能量计算更加精确,将Kohn-Sham单电子态平面波展开的截断能设置为520 eV,并选用OTFG超软赝势(ultrasoft pseudopotentials)来描述电子与离子芯之间的相互作用。在计算表面结构时,并未固定某一层的金属原子,而是选择让所有金属原子放开弛豫,虽然会增大计算量,但是计算结果会更接近实际情况。收敛标准设置为:最大原子位移为2.0×10-4nm,原子间最大作用力为0.5 eV/nm,总能量最大变化为每个原子2.0×10-5eV,自洽场迭代的收敛标准为每个原子2.0×10-6eV。由于镍和铁都是过渡金属,因此在计算过程中需要考虑自旋极化。Fermi拖曳效应设置为0.1 eV。针对所采用的p(2×2)超胞模型,这一周期表面的运算采用了8×8×1的布里渊区网格,并采用Monkhorst-Pack方案自动产生的不可约k点作自洽计算。对于体相材料,k点网络选用8×8×8。过渡态搜索方法为Complete LST-QST方法,过渡态搜索计算中所设定的收敛标准为(4.359 748 2±0.000 002 6)×10-19J/nm。
2.2 吸附能、共吸附能、反应能垒和反应能的定义式
单一吸附质吸附能的定义式为:
Qads=Qs+Qf-Qs+f
(1)
式中Qads——吸附能,eV
Qs——金属表面的总能量,eV
Qf——自由吸附质的总能量,eV
Qs+f——吸附质吸附在金属表面后的总能量,eV
如果Qads小于0,表示吸附过程为吸热反应;如果Qads大于0,表示吸附过程为放热反应。吸附能的绝对值越大则吸附越强。
定义2种吸附质在金属表面的共吸附能为:
Qco-ads=Qs+Qf,A+Qf,B-Qs+A+B
(2)
式中Qco-ads——共吸附能,eV
Qf,A、Qf,B——自由吸附质A、B的能量,eV
Qs+A+B——吸附质A和B共同吸附在金属表面后的总能量,eV
正反应能垒Qact,f、逆反应能垒Qact,b及反应能ΔQ的定义式为:
Qact,f=QTS-Qr
(3)
Qact,b=QTS-Qp
(4)
ΔQ=Qp-Qr
(5)
式中Qact,f——正反应能垒,eV
QTS——过渡态能量,eV
Qr——反应物能量,eV
Qact,b——逆反应能垒,eV
Qp——生成物能量,eV
ΔQ——反应能,eV
如果ΔQ小于0,表示为放热反应;如果ΔQ大于0,表示为吸热反应。
3 结果与讨论
3.1 CHx(x=0~4)和 H在Ni3Fe(111)表面上的吸附
甲烷在金属表面解离的产物是CHx(x=0~3)和H。对CHx(x=0~4)和H这6种吸附质,通过计算,得到不同吸附质在不同高对称吸附位的吸附能。吸附能的绝对值越大,吸附越强,吸附构型越稳定。通过比较某吸附质在不同高对称吸附位的吸附能,可以得到该吸附质在金属表面的最稳定吸附构型。
经计算,CHx(x=0~4)和H在Ni3Fe(111)表面最稳定吸附构型的吸附位和吸附能见表1,最稳定吸附构型见图3~8。图3~8中,紫色球为Fe原子,蓝色球为Ni原子,白色球为H原子,灰色球为C原子。

图3 CH4在Ni3Fe(111)表面上最稳定吸附构型
CHx(x=0~4)和H在Ni(111)表面最稳定吸附构型的吸附位和吸附能见表2,结合表1和表2可以看出,CHx(x=0~4)在Ni(111)和Ni3Fe(111)表面最稳定吸附构型的吸附能由大到小的顺序均是C、CH、CH2、CH3、CH4。此外,可以发现CH4在上述2个表面中的吸附能都很小,这是因为甲烷是饱和分子,构型十分稳定,而前人在实验上也证明了CH4在过渡金属表面的吸附能相当小,可以忽略不计[15-16],因此CH4的吸附过程可以认为是物理吸附[17]。

表1 CHx(x=0~4)和H在Ni3Fe(111)表面最稳定吸附构型的吸附位和吸附能

图4 CH3在Ni3Fe(111)表面上最稳定吸附构型

图6 CH在Ni3Fe(111)表面上最稳定吸附构型
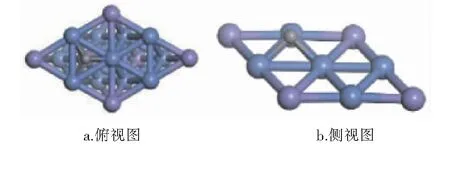
图7 C在Ni3Fe(111)表面上最稳定吸附构型
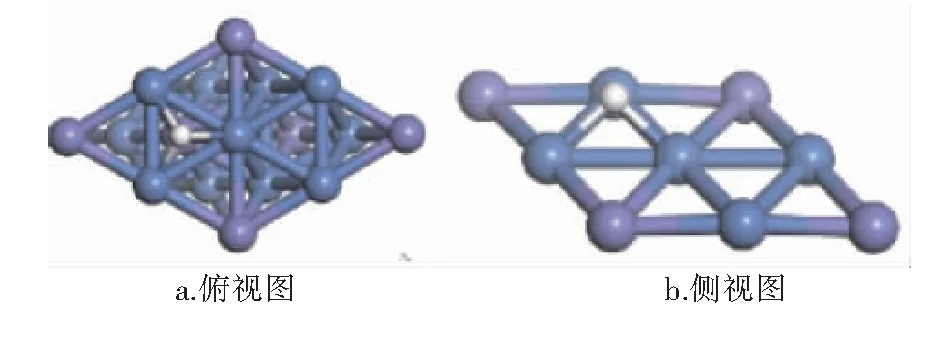
图8 H在Ni3Fe(111)表面上最稳定吸附构型
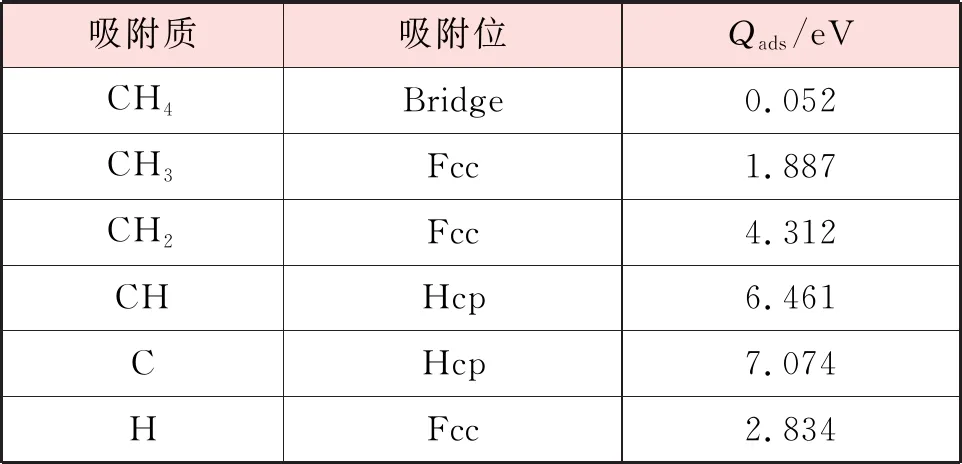
表2 CHx(x=0~4)和H在Ni(111)表面最稳定吸附构型的吸附位和吸附能
3.2 CHx(x=0~3)与H在Ni3Fe(111)表面上的共吸附
为了确定甲烷在金属表面解离过程的反应路线,需要探讨CHx(x=0~3)与H在金属表面的共吸附。通过计算,可以得到CHx(x=0~3)与H在金属表面不同高对称吸附位的共吸附能。共吸附能的绝对值越大,则吸附越强,该吸附构型越稳定。
CHx(x=0~3)与H在Ni(111)和Ni3Fe(111)表面上最稳定共吸附构型的吸附位和共吸附能见表3和表4。可以看出,CHx(x=0~3)与H在上述2个表面上的共吸附能都随x减小而增大,这与CHx(x=0~4)单吸附在这2个表面上的吸附能变化趋势一致。
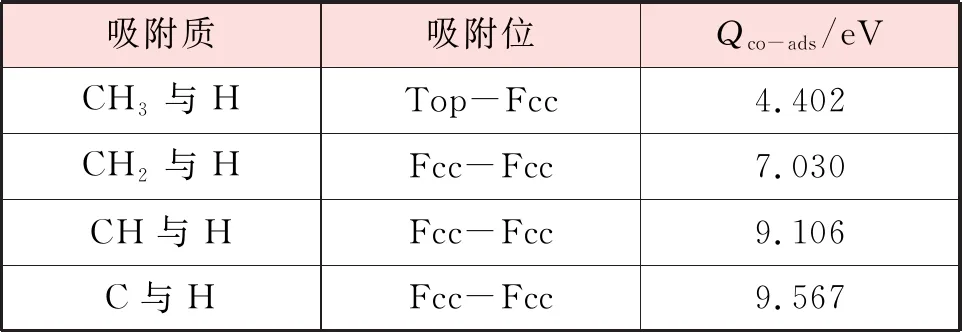
表3 CHx(x=0~3)与H在Ni(111)表面最稳定共吸附构型的吸附位和共吸附能

表4 CHx(x=0~3)与H在Ni3Fe(111)表面最稳定共吸附构型的吸附位和共吸附能
CHx(x=0~3)与H在Ni3Fe(111)表面上最稳定共吸附构型见图9~12。紫色球为Fe原子,蓝色球为Ni原子,白色球为H原子,灰色球为C原子。

图9 CH3与H在Ni3Fe (111)表面上最稳定共吸附构型
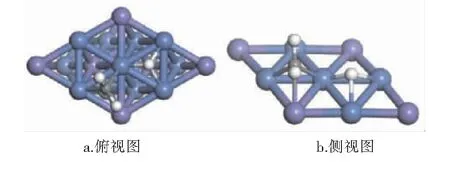
图10 CH2与H在Ni3Fe (111)表面上最稳定共吸附构型

图11 CH与H在Ni3Fe (111)表面上最稳定共吸附构型
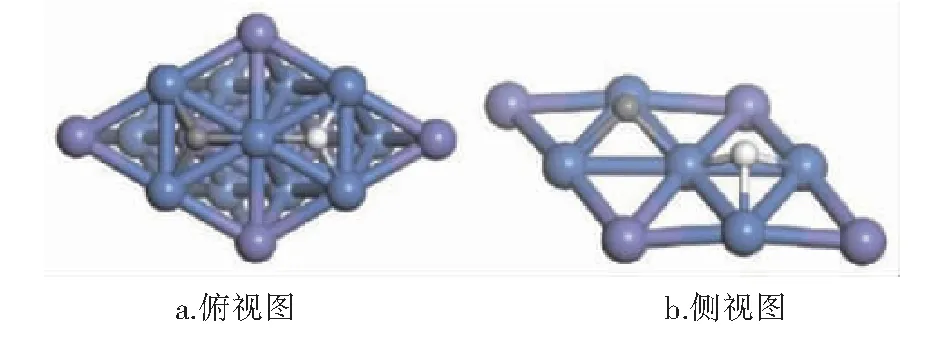
图12 C与H在Ni3Fe (111)表面上最稳定共吸附构型
3.3 CH4的脱氢过程
Wang等人[18]研究的甲烷逐步脱氢解离过程如下(TS1、TS2、TS3、TS4分别代表过渡态1、2、3、4):
CH4→TS1→CH3+H→CH3
CH3→TS2→CH2+H→CH2
CH2→TS3→CH+H→CH
CH→TS4→C+H→C
基于前文计算所得到的CHx(x=1~4)单吸附稳定构型以及CHx-1(x=1~4)与H共吸附稳定构型,对在Ni(111)以及Ni3Fe(111)表面的逐步脱氢反应进行了模拟。在每一步反应中,将对应吸附质的稳定吸附构型作为反应物和生成物。各步反应的反应能垒和反应能见表5和表6。观察甲烷在Ni(111)和Ni3Fe(111)表面上的各步解离反应的反应能,均有1个步骤为放热反应(CH2→CH+H),但相比于Ni(111)表面,CH2解离在Ni3Fe(111)表面上的放热更多。因此在Ni3Fe(111)表面甲烷的解离反应更容易发生。
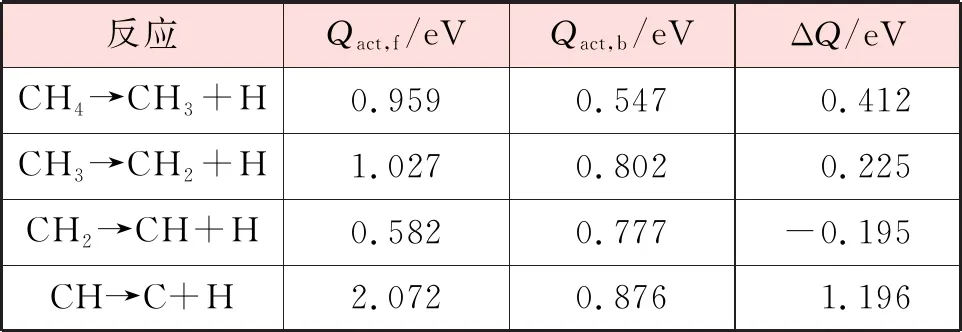
表5 CH4在Ni(111)表面逐步脱氢的正反应能垒、逆反应能垒和反应能
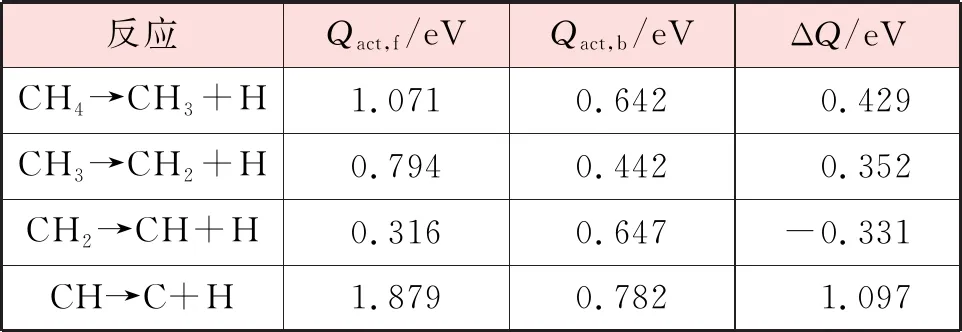
表6 CH4在Ni3Fe(111)表面逐步脱氢的正反应能垒、逆反应能垒和反应能
在Ni(111)和Ni3Fe(111)表面,CH脱氢反应具有最大的正反应能垒,因此它是反应的速率控制步骤。此外,CHx(x=1~3)在Ni(111)表面解离的正反应能垒大于Ni3Fe(111)表面,这表明铁的掺杂有利于CHx(x=1~3)物种在Ni基催化剂表面的解离,这一结论与前人的理论计算[19-20]和实验结果[21]一致。然而,CH4在Ni3Fe(111)表面解离的正反应能垒大于Ni(111)表面,这表明铁的掺杂会阻碍CH4的初步解离,这与Kim等人[22]的研究结果一致。为了更直观地对甲烷在Ni(111)和Ni3Fe(111)表面的解离过程进行比较,甲烷在2个表面逐步脱氢过程的能量变化见图13。在不考虑催化剂失活等因素的情况下,从图13中可以看出,甲烷在Ni3Fe(111)表面完全解离的整体正反应能垒大于Ni(111)表面,这说明Fe的掺入对于甲烷的解离是有利的。
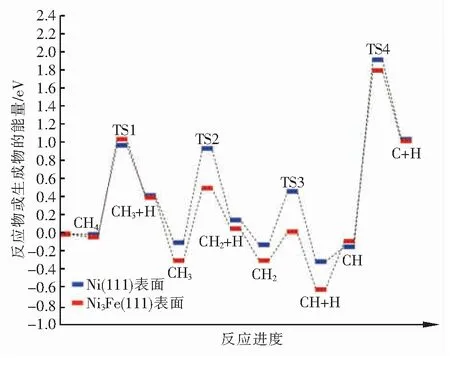
图13 CH4在Ni3Fe(111)和Ni(111)表面逐步脱氢过程的能量变化
3.4 甲烷解离各步基元反应的速率常数计算
为了进一步研究不同温度下甲烷在Ni3Fe(111)表面脱氢的机理,验证Fe的掺杂对于催化剂性能的提升,计算了298.15 K、500.15 K、700.15 K、973.15 K、1 023.15 K温度下甲烷解离各步基元反应的速率常数,见表7。比较速率常数可以发现,无论在Ni(111)表面还是在Ni3Fe(111)表面,CHx(x=1~4)物种解离难度由难到易依次为CH、CH4、CH3、CH2。速率常数的计算结果验证了第3.3节中通过反应能垒结果所得出的结论。
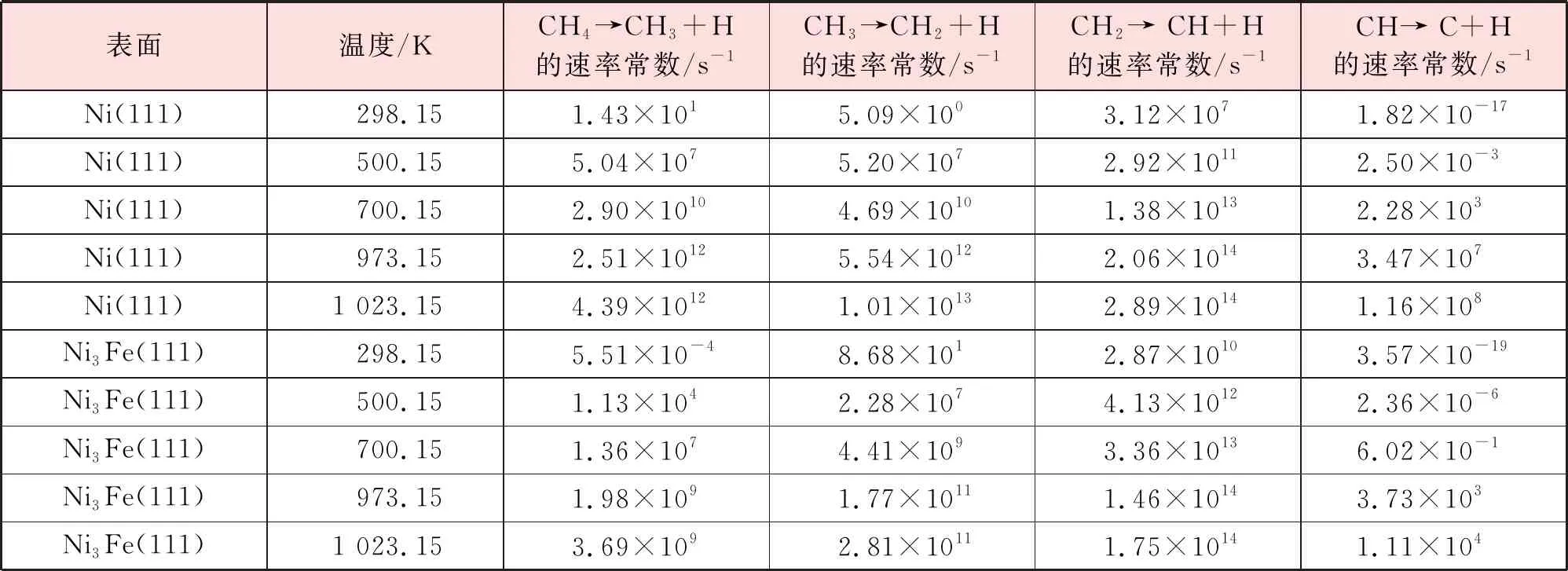
表7 不同温度下CH4在Ni(111)和Ni3Fe(111)表面脱氢反应的速率常数
4 结论
① 通过计算,得到甲烷脱氢解离过程中在Ni3Fe(111)表面上各吸附质最稳定吸附构型和各组吸附质最稳定共吸附构型。
② 分析甲烷脱氢解离各步反应的反应能垒和反应能。从动力学角度分析,甲烷在Ni(111)和Ni3Fe(111)表面上解离的速度控制步骤都是CH的裂解,而CH在Ni(111)表面解离的正反应能垒大于在Ni3Fe(111)表面解离的正反应能垒,表明Fe的掺杂可以降低甲烷解离的反应能垒。
③ 从热力学角度分析,相比于Ni(111)表面,CH2解离在Ni3Fe(111)表面的放热更多,所以在Ni3Fe(111)表面甲烷解离反应更易发生。
④ 动力学和热力学结果均表明Fe的掺杂可以提高Ni基催化剂催化甲烷解离的催化活性。
猜你喜欢
军民两用技术与产品(2022年1期)2022-06-01
成都信息工程大学学报(2021年4期)2021-11-22
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
燃料化学学报(2021年5期)2021-06-02
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
山东工业技术(2018年16期)2018-09-26
北京航空航天大学学报(2017年10期)2017-04-20
少年科学(2015年7期)2015-08-13
航天返回与遥感(2014年4期)2014-07-31
