
皂荚皂苷元制备及纯化工艺研究
2022-11-23 11:51:24刘艳萍范定臣刘佳佳赵俊杰
中国农学通报 2022年28期
刘艳萍,王 莹,张 静,范定臣,刘佳佳,赵俊杰,薛 刚
(1河南省林业科学研究院,郑州 450008;2南阳理工学院河南省工业微生物资源与发酵技术重点实验室,河南南阳 473004;3河南投资集团有限公司,郑州 450008;4河南省弘富林药业有限公司,河南内乡 474350)
0 引言
皂荚(Gleditsia sinensis)是中国特有的豆科皂荚属生态经济树种,分布于国内19个省(市)、自治区。皂荚荚果中皂苷含量在30%以上,是已知的皂苷含量最高的树种[1-2],皂苷具有独特的生物活性[1,3-4],在食品、化妆品、医药等行业的应用不断扩大[1,5]。皂苷元是皂苷的重要酸解产物,与皂苷分类划分依据相同可将其分为甾体皂苷元和三萜皂苷元,甾体皂苷元(如薯蓣皂苷元)是许多甾体激素的重要前体物质,具有多种重要的药理作用[6-7];三萜皂苷元(如皂荚皂苷元)具有消炎、抑制肿瘤和抗病毒等多种作用[8-9],可应用于生物、医药、金属加工等领域,拥有广阔的发展前景。目前从植物中制备皂苷元的方法主要有常规酸水解法[10-13]、酶解法[14-15]、超临界CO2萃取法[16]和微生物法[17]等。而相较于薯蓣皂苷元较成熟的制备技术,皂荚皂苷制备皂苷元的技术则处于摸索阶段。Zhang等[4]利用波谱测定确定了4种皂荚皂苷。皂苷元不溶于水,微溶于甲醇,溶于乙醇、乙酸乙酯、三氯乙烯等有机溶剂,但是对于亲酯类的石油醚、正己烷等溶解性很低,并且大多数皂苷元有良好的白色结晶。三萜皂苷元可以分为四环三萜和五环三萜2类,五环三萜皂苷元具有一些优秀的生物活性[16-21]。有关皂荚皂苷元分离纯化的文献报道较少。本研究采取有机溶剂萃取和重结晶的方法对皂荚果皮皂苷元的纯化工艺进行探究,旨在为其工业化生产提供参考。
1 材料与方法
1.1 材料与仪器
1.1.1 主要试剂 硫酸、盐酸、甲醇、无水乙醇、二氯甲烷、三氯乙烯、乙酸乙酯、正己烷、冰醋酸、高氯酸、香草醛等均为分析纯,60%皂荚皂苷粗品由西安森冉生物工程有限公司提供。
1.1.2 主要仪器 7890A气相色谱仪,美国安捷伦科技公司;1260型高效液相色谱仪,美国安捷伦科技公司;LXJ-IIB低速离心机,上海安亭科学仪器厂;752N型紫外可见分光光度计,上海电分析仪器有限公司等。
1.2 皂荚皂苷及皂苷元含量的检测
1.2.1 HPLC法测定含量
(1)色谱条件。
DAD法色谱柱为ZORBAX SB-C18柱(150 mm×4.6mm,5 μm),流动相为甲醇-水(95:5,V/V),波长280nm,流速1 mL/min,进样量10 μL。
ELSD法色谱柱同DAD法,流动相为甲醇-水(95:5,V/V),流速1.0 mL/min,柱温30℃,进样量10 μL,蒸发光散射器漂移管温度80℃,载气流速1.4 L/min。
(2)样品检测。取适量皂苷、皂苷元标准品和皂荚皂苷粗品用甲醇溶解,配制成1 mg/mL的溶液,用高效液相色谱仪进行测定。
1.2.2 气相色谱法测定酸解产物中皂苷元含量
(1)色谱条件。色谱柱为HP-5(30 m×0.32 mm),进样量4 μL,进样口温度330℃,FID 330℃,载气流速2 mL/min,分流比10:1,柱温280℃,以10℃/min升至310℃保持12 min。
(2)样品检测。取适量酸解产品用甲醇溶解,配制成1 mg/mL的溶液,用气相色谱仪进行测定。根据气相色谱图,通过对皂苷元峰面积所占百分比大小进行比较,判断皂苷元含量。
1.3 皂荚皂苷酸解工艺的单因素试验
1.3.1 酸浓度的选择 分别称取5份皂荚皂苷粗品各1 g,按照1:30(m:v)的料液比加入1、1.5、2、2.5、3 mol/L的浓硫酸溶液或2、3、4、5、6 mol/L的浓盐酸溶液,于90℃下水浴振荡,酸解4 h,酸解液真空抽滤,滤渣干燥称重,测定酸解产品中皂苷元的含量。
1.3.2 酸解时间的选择 分别称取4份皂荚皂苷粗品各1 g,按照1:30(m:v)的料液比加入已选择出的最优浓度的浓硫酸溶液或浓盐酸溶液,于90℃下水浴振荡,酸解时间为3、4、5、6 h,酸解液真空抽滤,滤渣干燥称重,测定酸解产品中皂苷元的含量。
1.3.3 酸解温度的选择 分别称取4份皂荚皂苷粗品各1 g,按照1:30(m:v)的料液比加入已选择出的最优浓度浓硫酸溶液或浓盐酸溶液,于80、85、90、95℃下水浴振荡,按选出的最优时间进行酸解,酸解液真空抽滤,滤渣干燥称重,测定酸解产品中皂苷元的含量。
1.3.4 料液比的选择 分别称取4份皂荚皂苷粗品各1 g,按照1:20(m:v)、1:30(m:v)、1:40(m:v)、1:50(m:v)的料液比加入已选择出最优浓度的浓硫酸溶液或浓盐酸溶液,于最佳温度下水浴振荡,按选出的最优时间进行酸解,酸解液真空抽滤,滤渣干燥称重,测定酸解产品中皂苷元的含量。
1.3.5 正交试验 根据酸解工艺条件对产物中皂苷元含量的影响试验,以酸解温度、酸种类、酸浓度、酸解时间4个因素设计正交试验,优化皂荚皂苷酸解工艺参数。
1.4 皂苷元萃取剂的选择
取3 g 60%皂荚皂苷原料,加入90 mL 5 mol/L盐酸,在90℃下酸解1 h,过滤得黑褐色固体为皂苷元粗品。分别加入90 mL甲醇、乙酸乙酯、二氯甲烷、三氯甲烷、三氯乙烯,过滤。观察现象。
1.4.1 萃取剂加量的选择 取3 g 60%皂荚皂苷原料,加入90 mL 5 mol/L盐酸,酸解1 h,过滤,取滤饼在60℃下烘干1 h,分别加入30、45、60、75、90 mL的溶剂,搅拌5 min,过滤,取滤液,旋干,回收溶剂,将所得固体放于60℃下烘干1 h,称重,待测。
1.4.2 萃取时间的选择 取3 g 60%皂荚皂苷原料,加入90 mL 5 mol/L盐酸,酸解1 h,过滤,取滤饼在60℃下烘干1 h,加入60 mL的溶剂,分别搅拌10、30、50、70、90 min后过滤,取滤液回收溶剂,将所得固体放于60℃下烘干1 h,称重。
1.4.3 萃取次数的选择 取3 g 60%皂荚皂苷原料,加入90 mL 5 mol/L盐酸,酸解1 h,过滤,取滤饼在60℃下烘干1 h,加入60 mL的溶剂,分别分成1、2、3次萃取,单次萃取时间为5 min。将所得滤液回收,将固体置于60℃下烘干1 h,称重。
1.4.4 皂苷元萃取纯化的正交试验 设立2、3、4、5 mol/L 4组不同的酸浓度,硫酸、盐酸2种不同酸种类,酸解时间分别为3、6 h,分别加入60 mL的二氯甲烷和三氯乙烯萃取剂。检验萃取前后产物含量差别。
1.5 皂苷元重结晶溶剂选择
溶剂,调整体系的极性和皂苷元的溶解性达到重结晶目的,测定所得重结晶产物中皂苷元的含量,确定最佳的重结晶剂。
1.6 数据分析
采用SPSS分析软件对试验结果进行数据处理与分析。
2 结果与分析
2.1 皂荚皂苷元检测条件的确定
分别向产物中加入三氯乙烯、甲醇、乙醇、丙酮,尽量使产物完全溶解,加入水或石油醚等皂苷元不溶的
2.1.1 HPLC法 使用HPLC-DAD法观察皂苷元和皂荚皂苷粗品出峰位置的差异。由图1~2可知,皂荚皂苷出峰时间早于皂苷元,皂苷在转化为皂苷元时出峰时间会向后推移,ELSD峰型好于DAD图谱。
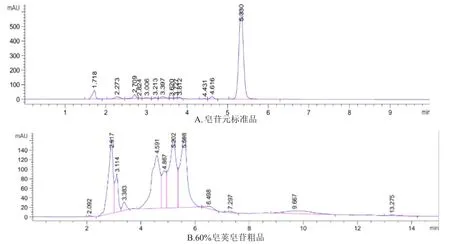
图1 DAD图谱
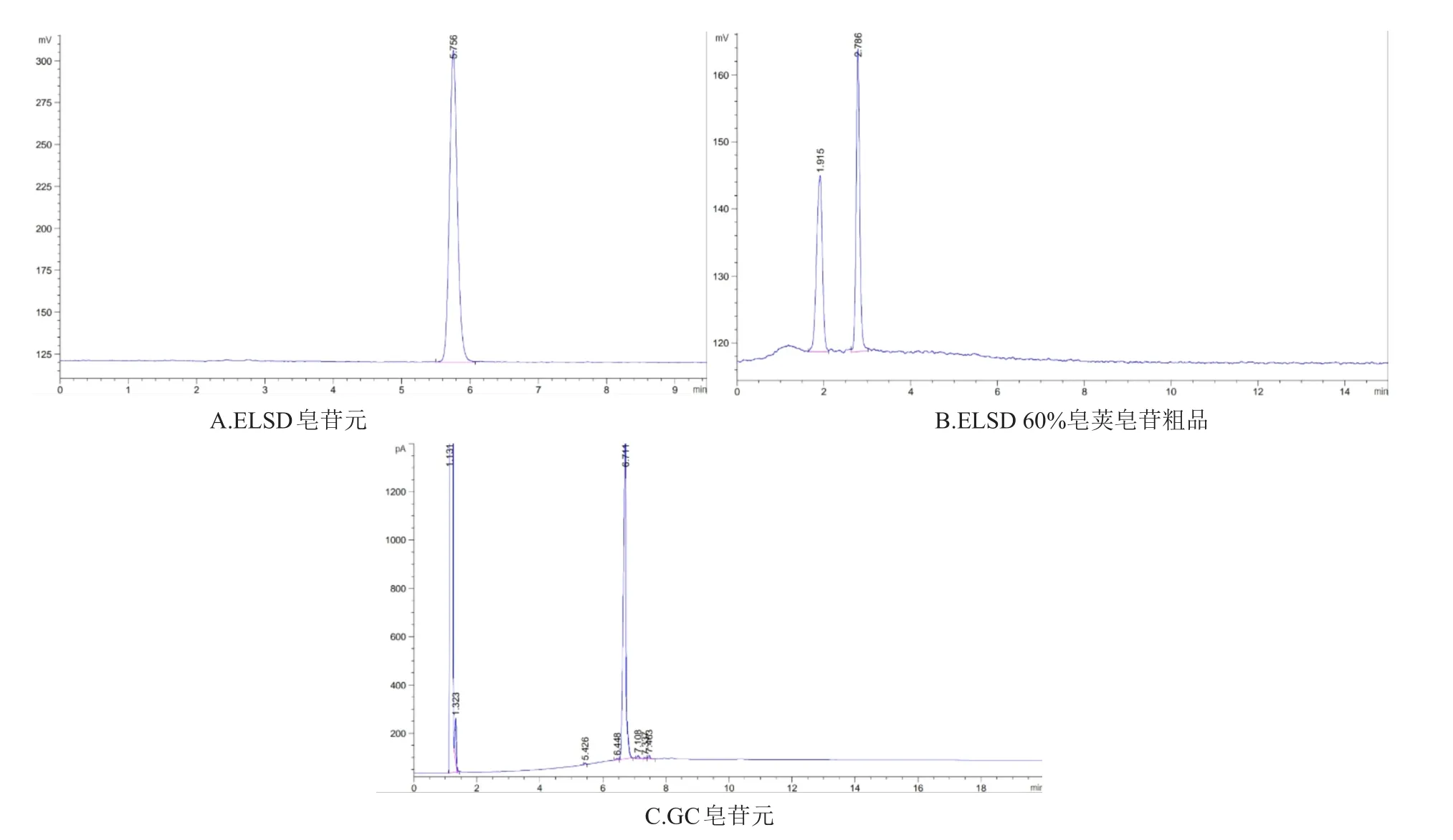
图2 ELSD和气相图谱
2.1.2 气相色谱法 取酸解的产物用气相色谱法进行检测,图谱见图2C:气相色谱法灵敏度高,能够检测出酸解出的微量皂苷元,还能将不同结构的皂荚皂苷元有效分离,因此选用气相色谱法检测皂荚皂苷元含量。
2.2 皂荚皂苷酸解工艺的单因素分析
2.2.1 最佳酸浓度的确定 料液比1:30,酸解温度为90℃,酸解4 h,气相色谱法对酸解产物的检测结果见表1。

表1 不同酸浓度硫酸和盐酸酸解产物的检测结果
硫酸浓度在1、2 mol/L范围内酸解产生皂荚皂苷元a(三萜皂苷元)变化比较大,2.5、3 mol/L范围变化不大;但是在1、1.5、2、2.5 mol/L的浓度均无法产生皂荚皂苷元b(甾体皂苷元),3 mol/L开始产生皂荚皂苷元b。
盐酸浓度在2、3、4、5 mol/L范围内对产生的皂荚皂苷元a和皂荚皂苷元b含量影响不大,浓度达到6 mol/L有下降趋势,酸浓度增加对皂荚皂苷元a和皂荚皂苷元b的产生有抑制作用。
综上,硫酸浓度3 mol/L、盐酸浓度5 mol/L的工艺条件更佳。
2.2.2 最佳酸解时间的确定 选取3 mol/L的硫酸溶液或2 mol/L的盐酸溶液以1:30的料液比,酸解温度90℃,酸解时间为3、4、5、6 h的条件进行酸解,气相色谱法对酸解产物的检测结果见表2。
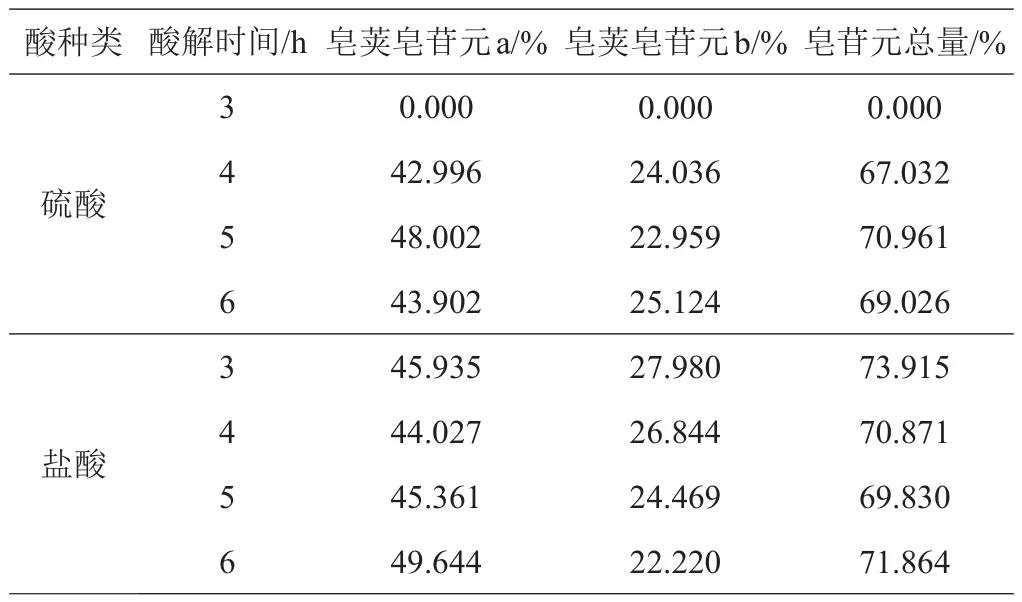
表2 不同时间硫酸和盐酸酸解产物的检测结果
硫酸酸解生成皂苷元总量和皂荚皂苷元a随着时间的增加而增加,在5 h时达到最大,6 h开始减少;生成皂荚皂苷元b随着时间的增加而增加。盐酸酸解生成皂荚皂苷元a随着时间的增加而缓慢增加,整体变化不大;生成皂荚皂苷元b随着时间的增加而减少;生成皂苷元总量随时间增加而减少,总体变化不大。确定硫酸酸解时间5 h、盐酸酸解时间3 h的工艺条件更佳。
2.2.3 最佳酸解温度的确定 选用3 mol/L的硫酸溶液或2 mol/L的盐酸溶液以1:30的料液比,酸解温度分别为80、85、90、95℃,酸解时间为4 h的条件进行酸解,气相色谱法对酸解产物的检测结果如表3所示。
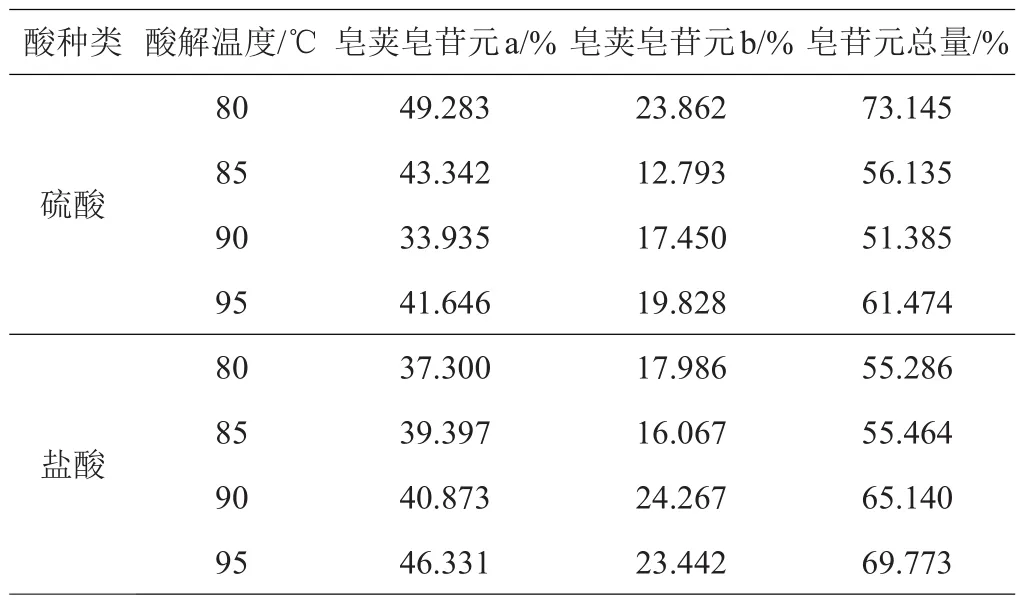
表3 不同温度硫酸和盐酸酸解产物的检测结果
随着温度的增加,硫酸酸解生成皂荚皂苷元含量减少,盐酸酸解生成皂苷元总量增加。确定硫酸酸解温度80℃、盐酸酸解温度95℃的条件更佳。
2.2.4 最佳酸解料液比的确定 选用3 mol/L的硫酸溶液或2 mol/L的盐酸溶液以1:20、1:30、1:40、1:50的不同料液比,酸解温度90℃,酸解时间4 h的条件进行酸解,气相色谱法对酸解产物的检测结果见表4。不同料液比酸解时,选择硫酸料液比1:20、盐酸料液比1:30的工艺条件更佳。
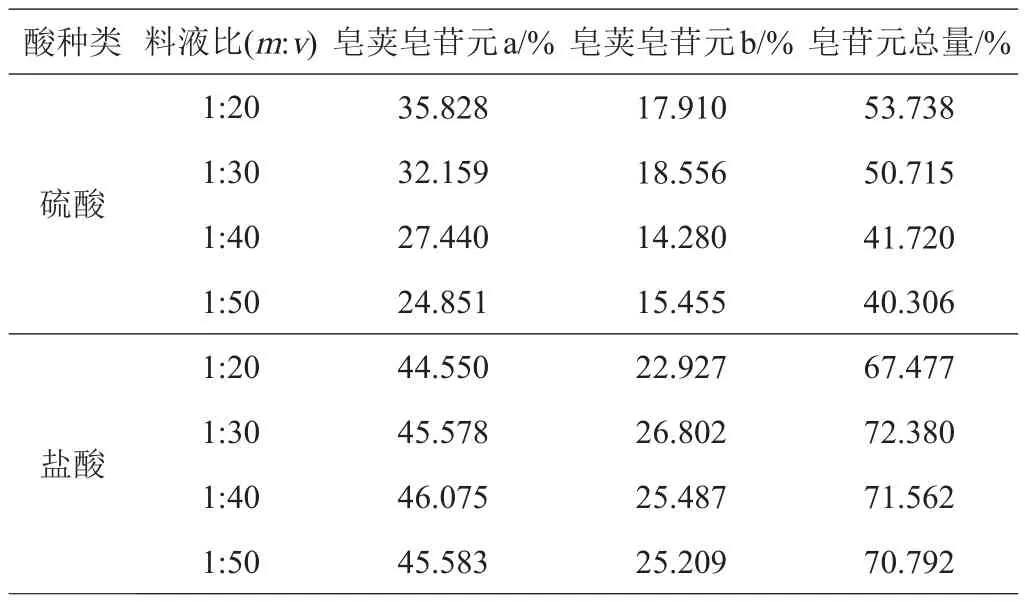
表4 不同料液比硫酸和盐酸酸解产物检测结果
2.3 皂苷元纯化工艺分析
2.3.1 皂苷元萃取剂的选择 皂苷元在水和石油醚中几乎不溶,在乙醇中部分溶解,二氯甲烷中几乎全溶。加入甲醇中时有大量沉淀。乙醇在加至6 mL时皂苷元完全溶解。乙酸乙酯在加至5 mL皂苷元完全溶解。由此可知,氯代烃(氯仿、二氯甲烷等)、乙酸乙酯等可以成为皂苷元提取剂。将黑褐色皂苷元粗品加入萃取剂后,过滤。所得甲醇、乙酸乙酯滤液为均黑色溶液;二氯甲烷滤液为红褐色溶液;三氯甲烷为黄褐色溶液;三氯乙烯为黄色溶液。因三氯乙烯溶液颜色最浅,色素含量少,且溶解效果好,最终确定萃取溶剂为三氯乙烯。
2.3.2 萃取剂用量确定 由于在液相色谱上出峰位置较为难分开,故采用气相色谱法做初步的纯化检验。纯化效果检验采用峰面积比较法。三氯乙烯萃取所纯化的含量没有随用量大小发生明显的改变,结果见表5。

表5 萃取剂的用量对产品质量影响
从整体趋势看,在萃取剂用量达到60 mL时已经到极限,后续增加萃取剂用量不会增加萃取效果。
2.3.3 萃取时间的确定 萃取剂萃取时间对产品质量影响结果如表6所示,产品质量变化为0.26~0.28 g,整体变化趋势平缓,萃取时间对萃取效果影响不大。
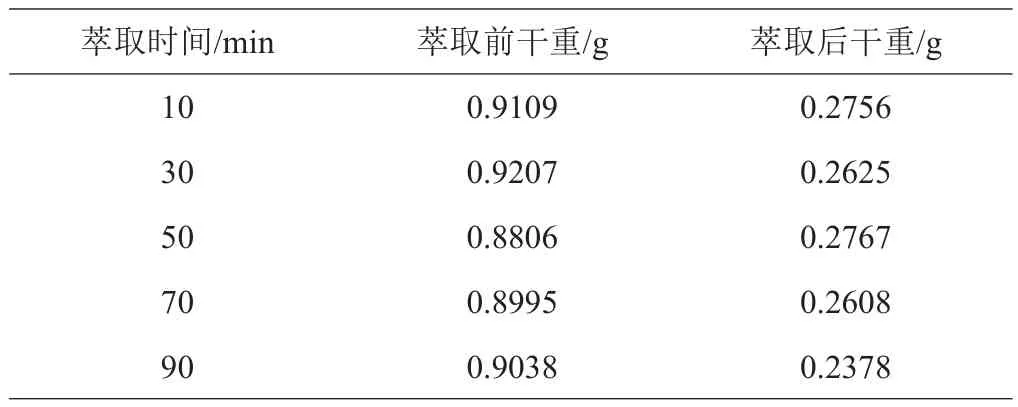
表6 萃取剂的萃取时间对产品质量影响
2.3.4 萃取次数的确定 萃取1次后干重为0.2660 g,萃取2次后干重为0.2220g,萃取3次后干重为0.2126g。萃取次数越多,所得皂苷元越少,认为在滤纸上损失的产品过多。所以三氯乙烯萃取皂苷元仅用1次萃取即可。
从单因素试验结果来看,萃取时间对萃取效果影响不大,从酸解产品中萃取用质量体积比20倍的三氯乙烯萃取1次即可。
2.4 重结晶溶剂的选择
利用单因素试验所得萃取过后的产品进行重结晶的溶解性试验。(1)100倍甲醇50℃热溶固体尚未完全溶解,溶剂用量过大,工业化困难,不再做考虑。(2)皂苷元几乎不溶于正己烷,向19 mL三氯乙烯萃取液中加入3 mL正己烷,出现白色沉淀,静置10 min后白色粉末结成固体,表面覆盖一层黄色物质,此方法需要快速过滤,对于工业生产较为苛刻,此种方法不再做考虑。(3)加入无水乙醇20倍,固体完全溶解,逐步加水调整极性。在80%乙醇溶液时,出现稳定的白色固体,当调整到60%乙醇溶液时出现大量白色固体。(4)丙酮重结晶也会出现与乙醇相同的现象,但丙酮重结晶的白色粉末更为细小,在滤纸上难以存留。选择60%乙醇作为重结晶试剂。
2.5 正交试验
2.5.1 皂苷酸解正交试验 基于皂荚皂苷酸解工艺条件的单因素试验数据及分析,选择酸解温度(A1=80℃,A2=85℃,A3=90℃,A4=95℃)、酸种类(B1=硫酸,B2=盐酸)、酸浓度(C1=4 mol/L,C2=6 mol/L)和酸解时间(D1=4 h,D2=6 h)4个影响因素采用L8(27)正交试验表进行试验,结果见表7。
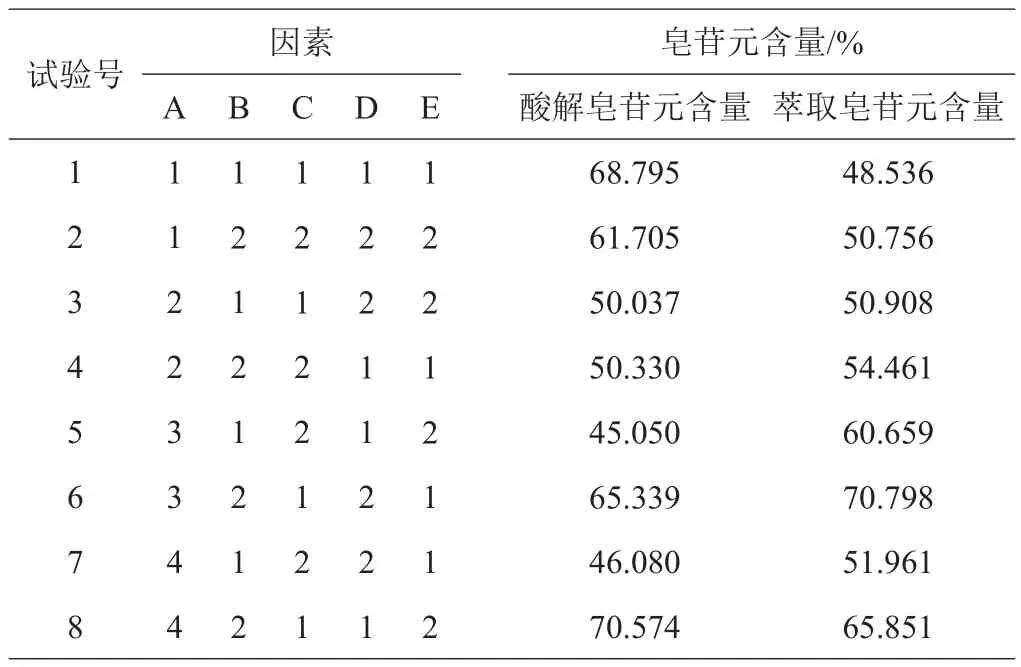
表7 皂苷酸解正交试验方案及结果
2.5.2 萃取纯化正交试验 同上,采用L8(27)正交试验考察不同的酸浓度(A1=2 mol/L,A2=3 mol/L,A3=4 mol/L,A4=5 mol/L)、萃取剂种类(B1=二氯甲烷,B2=三氯乙烯)、酸种类(C1=盐酸,C2=硫酸)、酸解时间(D1=3 h,D2=6 h)对纯化效果的影响,检验萃取前后产物含量差别。正交试验产品通过气相色谱比对全部产物纯化前和纯化后的含量。
对表7数据进行方差分析得表8。

表8 萃取纯化正交试验方差分析
皂苷元酸解正交试验结果表明,C因素对结果的影响到达显著水平,B因素接近显著,应控制在优水平。A、D可以选择任意水平,最优水平组合为A1B2C1D1,即盐酸浓度为4 mol/L,酸解温度为80℃,酸解时间为4 h,料液比为1:30。
萃取纯化正交试验结果表明,最优水平组合为A3B2C1D1,即4 mol/L盐酸酸解3 h,用三氯乙烯萃取。
3 结论
皂苷是一种苷类中结构比较复杂的化合物,存在于许多中草药与陆生植物中,也少量存在于海星和海参等海洋生物中。皂荚荚果中皂苷含量在30%以上,是已知的皂苷含量最高的植物。皂苷具有表面活性和多种重要的生物活性,皂苷转化为皂苷元后,可直接用于制剂药物,广泛应用于利尿、抗病毒、降血糖、防治心脑血管疾病等,也可以作为原料药进一步合成新药,具有很广阔的应用空间[22]。本试验以皂荚皂苷粗品为原料,在单因素分析的前提下设计正交试验,考察酸解温度、酸种类、酸浓度、酸解时间4个因素的影响,优化皂荚皂苷酸解工艺参数,筛选出最佳工艺为盐酸浓度4 mol/L,酸解时间4 h,酸解温度80℃,料液比1:30(m:v)。通过有机溶剂萃取法和重结晶法对其酸水解后的皂苷元进行纯化,确定最佳的萃取剂为三氯乙烯,料液比为1:20(m:v),萃取时间在90 min内变化不大,萃取1次,采用60%乙醇重结晶,得到的总皂苷元纯度可达76.31%。研究结果可为皂荚皂苷元工业化的生产提供理论依据。
4 讨论
查阅相关的文献研究发现,关于薯蓣皂苷元的制备研究颇多,工业生产技术相对成熟,但是从皂荚中提取皂苷和制备皂苷元的研究较少,薯蓣皂苷元制备工艺对皂荚皂苷元的提取具有一定的借鉴意义。在有机试剂纯化皂荚皂苷元的工艺优化中,溶解性试验结果显示石油醚不适合作为皂荚皂苷元的萃取剂,这与部分文献[23-25]报道的石油醚提取薯蓣皂苷元结果相悖。
由于目前尚无皂荚皂苷元标准品,本研究尝试采用气相色谱法和液相色谱法对皂荚皂苷水解产物进行分析检测,为皂苷元的结构分析和含量检测奠定基础。首先对皂荚皂苷酸解产物进行气相色谱检测,谱图中出现3个随着纯化工艺改变,含量随之出现增减变化的组分峰。通过DAD检测器发现皂苷元产品在210 nm与280 nm处有紫外吸收,且210 nm处略强。通过与文献比对,齐墩果烷类型含有环内孤立双键时在205~215 nm处有最大吸收,结构中含有羰基在285 nm处有紫外光吸收,结合文献报道[6],判断皂荚皂苷水解产物中含有齐墩果烷类型的皂苷元,其结构中有一双键、一羰基。ELSD-LC检测重结晶脱色处理后的皂苷元产品,确定其中有3个皂苷元组分的液相色谱峰,为后续皂荚皂苷元的结构确认及含量测定方法研究提供参考。
猜你喜欢
散文选刊·下半月(2024年3期)2024-05-01 09:02:48
绿色天府(2023年10期)2023-12-05 09:30:32
中成药(2017年9期)2017-12-19 13:34:31
中成药(2017年9期)2017-12-19 13:34:28
幸福·婚姻版(2016年1期)2016-03-07 12:52:12
华东理工大学学报(自然科学版)(2015年1期)2015-11-07 09:15:33
医学研究杂志(2015年8期)2015-06-22 14:00:57
癌变·畸变·突变(2015年3期)2015-02-27 06:15:10
中华皮肤科杂志(2014年4期)2014-12-19 12:56:00
中华皮肤科杂志(2014年4期)2014-12-19 12:55:43
