
表面水热碳层对磁性NiFe2O4八面体光催化活性的影响
2022-11-15 09:34:52郭彪赵晨灿刘芯辛于洲周丽景袁宏明赵震
高等学校化学学报 2022年11期
郭彪,赵晨灿,刘芯辛,于洲,周丽景,袁宏明,赵震
(1.沈阳师范大学化学化工学院,沈阳 110034;2.吉林大学化学学院,无机合成与制备化学国家重点实验室,长春 130012)
随着现代工业的快速发展,越来越多有毒有害的印染废水被释放到环境中,危害着人类的健康,环境污染已经成为全球普遍关注的焦点.水是人们赖以生存的物质,水污染的治理显得尤为重要.目前,废水中染料处理的方法有吸附沉淀法、生物处理法、膜技术等,但这些方法操作成本较高,容易产生二次污染[1].而半导体光催化技术可以利用清洁的太阳光降解有害染料,此方法清洁环保、操作简便、条件温和,受到众多科学家的重视[2].
目前,已经发展了众多半导体光催化剂,如TiO2[3],ZnO[4],Fe2O3[5],CdS[6],BiVO4[7],g-C3N4[8]等.然而,将这些光催化剂应用于实际生产仍然面临着很多挑战.首先,它们的可见光的吸收范围较窄,导致无法利用大部分的太阳光.其次,光生载流子的复合速率太快,导致较低的量子效率.最重要的是,在实际应用中要求用过的催化剂能从水中快速分离循环利用,否则就会导致二次污染,然而,大部分催化剂无法满足上述条件.为了解决这些问题,发现了很多高活性磁性半导体光催化剂(如NiFe2O4[9],ZnFe2O4[10],CuFe2O4[11],CoFe2O4[12]等).在这些磁性材料中,NiFe2O4因为其具有带隙(1.7 eV)较窄、成本低、化学稳定性好、合成简单、无毒无害及易分离等优点被广泛研究[13].但是,纯NiFe2O4的光催化活性较差,这是因为其光生载流子复合速度太快.很多研究小组通过耦合半导体构建异质结来提高NiFe2O4的光催化活性.已经报道的NiFe2O4基光异质结催化剂有Ag3PO4/NiFe2O4[14],NiFe2O4@TiO2[15],BiOI/NiFe2O4[16],NiFe2O4/g-C3N4[17]和NiFe2O4/Grapheme[18]等.
碳材料具有良好的导电性、生物相容性、价格低廉、经济环保、稳定性高等特性,在各个领域都有广泛的应用[19].碳材料在光催化中可以有效提高光生电子的迁移率,降低载流子的复合几率.将碳材料与NiFe2O4复合可改善其光催化活性.如Rigo等[20]通过微波辐射技术合成了铁酸镍/多层碳纳米管(NiFe2O4/MWCNT)复合材料.Zhao等[21]合成了铁酸镍/石墨烯(NiFe2O4/Graphene)纳米复合材料.虽然上述构筑的碳材料/NiFe2O4异质结一定程度上能增强NiFe2O4的光催化活性.但是,由于碳材料和NiFe2O4之间的接触面积有限,这会限制界面载流子的分离效率.利用水热碳层包覆半导体制备核壳结构,可有效增加碳材料和半导体之间的接触面积[22].但是用水热碳层包覆NiFe2O4制备核壳结构的异质结很少有文献报道,同时表面水热碳层的厚度对NiFe2O4光催化活性的影响也有待明确.
本文通过葡萄糖水热碳化法将碳包覆在NiFe2O4八面体上,通过调控葡萄糖的含量合成了一系列不同碳层厚度的NiFe2O4核壳八面体(NiFe2O4@C)复合材料,表征了其结构、组成、形貌,评价了其对亚甲基蓝降解的光催化性能和稳定性,并且探究了水热碳层厚度对NiFe2O4光生电荷性质及光催化活性的影响机制.研究结果有望为高活性半导体核/壳结构光催化剂的设计提供新的思路.
1 实验部分
1.1 试剂与仪器
九水合硝酸铁[Fe(NO3)3·9H2O]、六水合硝酸镍[Ni(NO3)2·6H2O]、氢氧化钾(KOH)、异丙醇、葡萄糖、亚甲基蓝(MB)、叔丁醇(TBA)、乙二胺四乙酸二钠盐(EDTA-2Na)、硝酸银(AgNO3)和对苯醌(BQ)均为分析纯,购自国药集团化学试剂有限公司;Nafion(5%,体积分数)购自西格玛奥德里奇有限公司.
Rigaku D-Max 2550型X射线粉末衍射仪(XRD,2θ=20°~80°,10°/min,日本Rigaku公司);INVIA型拉曼光谱仪(Raman,英国雷尼绍公司);VG Scienta R3000型X射线光电子能谱仪(XPS,英国VG Scienta公司);TGA Q500型热重分析仪(TGA,美国TA公司);U-4100型紫外-可见漫反射光谱仪(UV-Vis DRS,日本岛津公司);JSM-6700F型扫描电子显微镜(SEM,日本电子公司);Tecnai G2 S-Twin F20型200 kV透射电子显微镜(TEM,美国FEI公司);F-7100型荧光光谱仪(PL,HITACHI日立公司);CHI660Ed电化学工作站(上海辰华仪器有限公司);ICPOES730型电感耦合等离子体原子发射光谱仪(ICP-OES,美国安捷伦公司).
1.2 实验过程
1.2.1 NiFe2O4八面体的制备将7.6 mmol Fe(NO3)3·9H2O和3.8 mmol Ni(NO3)2·6H2O溶解在30 mL去离子水中,在室温下磁力搅拌2 h.将30 mL 2.0 mol/L的KOH溶液逐滴加入到上述溶液中,继续搅拌30 min,然后将混合溶液转移至90 mL内衬为聚四氟乙烯的反应釜中,并在180°C下水热反应22 h.待自然冷却至室温后,产物用磁铁分离,收集的沉淀物用乙醇和水各洗涤3次,并在80°C下干燥5 h,即得到NiFe2O4八面体.
1.2.2 NiFe2O4@C核壳八面体的制备以葡萄糖为碳源,通过水热法合成了NiFe2O4@C核壳八面体.首先,将一定量的葡萄糖加入到40 mL去离子水中,搅拌30 min.随后将3 mmol已制备的NiFe2O4加入到葡糖糖水溶液中,搅拌30 min后,将混合溶液转移至90 mL内衬为聚四氟乙烯的反应釜中,并在180℃的烘箱中放置22 h.冷却到室温后,产物用磁铁分离,收集的沉淀物用乙醇和水各洗涤3次,并在80℃下干燥5 h.最后,将产物在N2气保护下在管式炉中于300℃煅烧6 h,最终制备得到NiFe2O4@C核壳八面体.通过改变葡萄糖的量(1,3,9 mmol)来调控碳层的厚度,将制备的不同碳层厚度的样品分别记为NiFe2O4@C-1,NiFe2O4@C-3和NiFe2O4@C-9.
NiFe2O4八面体和NiFe2O4@C核壳八面体的制备过程见Scheme 1.
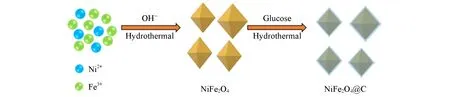
Scheme 1 Fabrication procedures of NiFe2O4 octahedron and NiFe2O4@C core-shell octahedron
1.2.3 光催化性能测试通过光催化降解MB来评价样品的光催化性能.将0.05 g催化剂分散到50 mL 10 mg/L的MB溶液中,然后置于250 mL石英反应器中.为了使实验达到吸附解析平衡,先在黑暗条件下搅拌2 h,之后打开300 W氙灯,光照期间一直通冷凝水,将温度控制为室温.之后每间隔30 min取一次混合溶液(3 mL),用磁铁将催化剂和溶液分离,溶液用紫外-可见分光光度计测试吸收光谱.
1.2.4 光电化学性能测试使用电化学工作站测试了不同样品的光电化学性能,工作电极的制备:将相同质量的4个样品(NiFe2O4,NiFe2O4@C-1,NiFe2O4@C-3,NiFe2O4@C-9)超声分散到2 mL含1%(体积分数)Nafion的异丙醇溶液中,然后涂在0.5 cm×0.5 cm的氧化铟锡(ITO)玻璃片上,干燥后制得工作电极.光电流测试使用300 W氙灯作为光源,0.5 mol/L Na2SO4溶液作为电解液,分别以碳棒和饱和甘汞电极作为对电极和参比电极.电化学阻抗谱(EIS)是在暗态下,外加10 mV偏压,0.1 Hz-100 kHz频率范围内进行测试.
2 结果与讨论
2.1 催化剂的表征
为了确定所制样品的组成,首先进行了XRD测试.如图1(A)所示,纯NiFe2O4和NiFe2O4@C均在2θ=30.3°,35.7°,43.4°,53.8°,57.4°,63.0°和74.6°处出现较强的衍射峰,这些峰与立方尖晶石NiFe2O4的标准卡片(PDF#54-0964)完美对应,说明制备的样品中都存在结晶度较高的NiFe2O4.在XRD谱图中没有观察到其它杂相的峰,说明所制样品为纯的NiFe2O4.另外,NiFe2O4@C只有纯的NiFe2O4的峰,没有观察到对应碳的衍射峰,这可能是因为碳的结晶度比较低,XRD无法检测到.值得注意的是,不同碳层厚度的NiFe2O4@C复合材料具有相同的XRD衍射峰强度,证明NiFe2O4的晶体结构在水热过程中没有被破坏.通过拉曼光谱表征来进一步确定样品中碳的存在.图1(B)显示了NiFe2O4和NiFe2O4@C的拉曼光谱,相对于纯NiFe2O4,所有NiFe2O4@C样品的拉曼光谱均在1330和1590 cm-1处存在两个明显的峰,1330 cm-1处的D峰表示存在无序或低对称的碳,1590 cm-1处的G峰表示存在sp2杂化石墨碳.图中显示D峰和G峰均较宽而且强度较弱,可能是因为石墨烯中存在无定型碳.此外,所有样品均在100~1000 cm-1范围内出现了一些峰,归属于NiFe2O4,这与NiFe2O4的研究结果一致[23~25].结合XRD和Raman表征结果,证明合成的复合材料包含NiFe2O4和碳.

Fig.1 XRD patterns(A)and Raman spectra(B)of NiFe2O4 and NiFe2O4@C composites
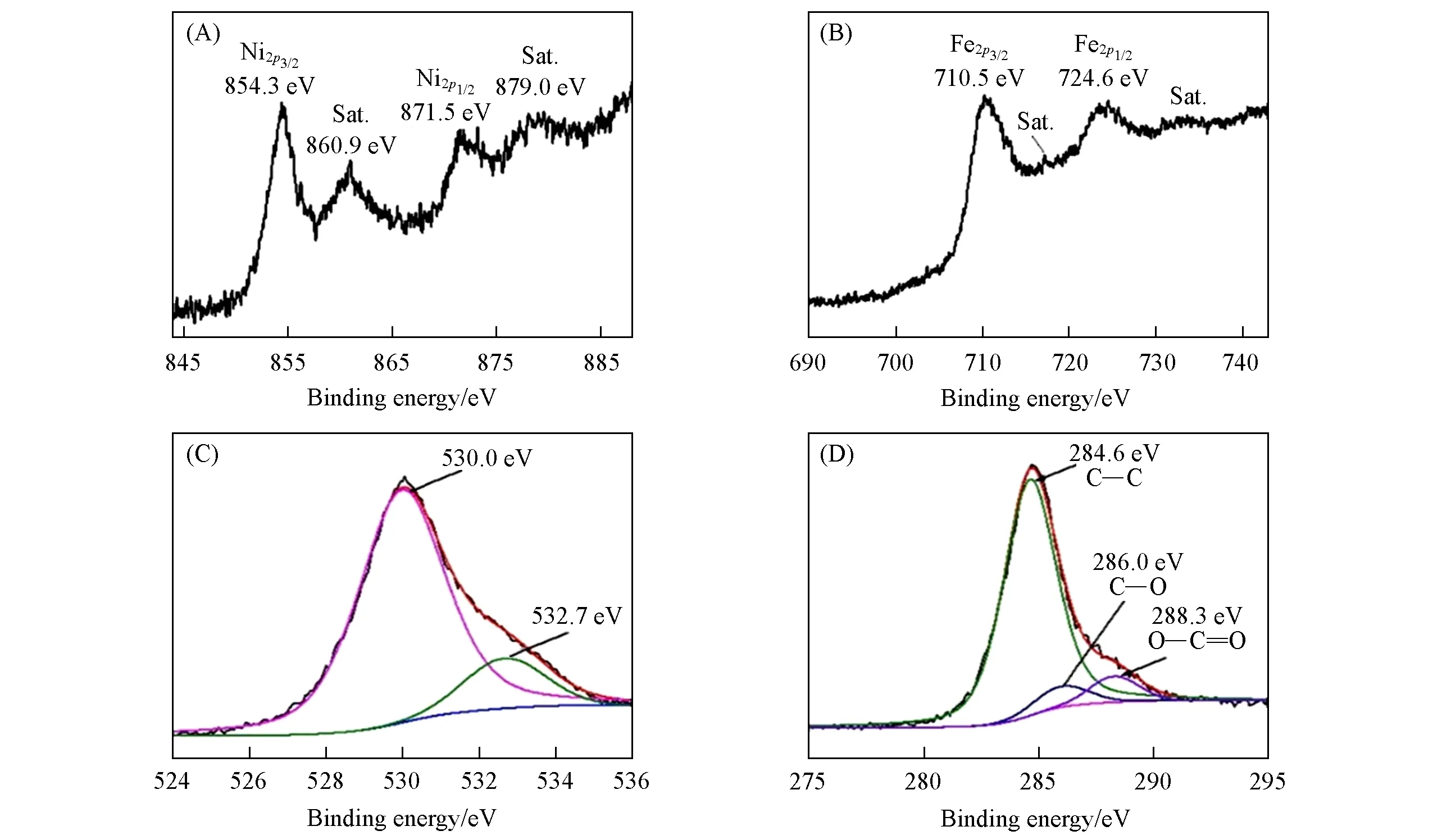
Fig.2 XPS spectra of Ni2p(A),Fe2p(B),O1s(C)and C1s(D)for the NiFe2O4@C-3
通过XPS来确定制备的NiFe2O4@C-3的元素组成和化学价态.图2(A)展现了Ni2p的XPS谱图,854.3和871.5 eV处的峰分别归属为Ni2p32和Ni2p12的自旋轨道,表明样品中存在Ni2+,860.9和879.0 eV处的峰与卫星峰有关[26].Fe2p的XPS谱图中710.5和724.6 eV处的峰分别对应于Fe2p32和Fe2p12的自旋轨道,谱图中也出现了两个小的卫星峰[图2(B)],表明制备的样品中存在Fe3+.O1s的XPS谱图中530.0 eV处的峰归属于典型的金属-氧键,532.7 eV处的峰归属于O缺陷[图2(C)],O缺陷包括表面吸附的OH-,O2-等.这些结果进一步证明了NiFe2O4@C中存在Ni2+,Fe3+,O2-.图2(D)中C1s的XPS谱图可以分为3个峰,分别为284.6,286.0和288.3 eV,它们对应于C—C,C—O和O—C=O键[26].XPS数据进一步证明制备的样品中存在Ni,Fe,O和C元素.
通过SEM和TEM表征了NiFe2O4和NiFe2O4@C的形貌.图3(A)~(D)展示了4个样品的SEM照片,从图中可以看出,NiFe2O4和NiFe2O4@C的尺寸大小不均匀,粒径大约在100~500 nm之间,但是它们的形状相对均匀,均为八面体.碳包覆NiFe2O4以后,其形貌没有发生变化,这与XRD表征结果相一致.但是,在SEM照片中很难清晰观察到碳层及其厚度,所以,需要通过TEM和高分辨率透射电子显微镜(HRTEM)进一步来表征其形貌结构.通过动态光散射测试了NiFe2O4和NiFe2O4@C的粒径分布,结果如图S1和表S1(见本文支持信息)所示,NiFe2O4和NiFe2O4@C复合材料的平均粒径相近.

Fig.3 SEM images of NiFe2O4(A),NiFe2O4@C-1(B),NiFe2O4@C-3(C)and NiFe2O4@C-9(D)
图4展示了NiFe2O4和NiFe2O4@C的TEM和HRTEM照片.进一步证实了NiFe2O4和NiFe2O4@C为八面体.相对于纯NiFe2O4[图4(A)],所有NiFe2O4@C八面体边缘明显有一层颜色较浅的碳层,形成了核壳结构[图4(C),(E)和(G)].从图4(B)的HRTEM照片可以清晰观察到纯NiFe2O4有3种晶格间距,分别为0.253,0.295和0.489 nm,分别对应于立方NiFe2O4的(311),(220)和(111)晶面,这表明制备的NiFe2O4有较好的结晶度[27].从图4(D),(F)和(H)可以看到,随着葡萄糖含量的增加,制备的NiFe2O4@C的碳层厚度也随之增加,NiFe2O4@C-1,NiFe2O4@C-3和NiFe2O4@C-9的碳层厚度分别为3.7,5.5和13.7 nm.因为碳层包覆的影响,NiFe2O4@C-1和NiFe2O4@C-9的HRTEM照片中只观察到了0.295 nm的晶格条纹,NiFe2O4@C-3的HRTEM照片中观察到了0.295和0.489 nm的晶格条纹.为了进一步确定碳层包裹在NiFe2O4表面,通过TEM表征获得了NiFe2O4@C-3各元素的X射线能谱(EDS)元素面扫图(Mapping).结果如图4(I)和(J)~(M)所示,可见,Ni,Fe,O和C元素均匀分布在NiFe2O4@C-3八面体表面.此外,C元素形成的八面体面积大于Ni,Fe,O,这进一步证明C层包覆在NiFe2O4八面体表面形成了核壳结构的异质结.总之,通过SEM和TEM可以证明制备的样品为NiFe2O4@C核壳八面体,NiFe2O4八面体作为核,碳层作为壳,彼此紧密接触形成了异质结结构,这有利于促使光生载流子的分离和传输.
为了研究所制样品的热稳定性和NiFe2O4@C复合材料中C的含量,在空气中,对NiFe2O4和NiFe2O4@C样品进行TGA分析.结果如图5所示,纯NiFe2O4的热稳定性较好,在30~800℃范围内仅损失1.8%.与纯NiFe2O4相比,所有NiFe2O4@C样品均表现出两个失重过程,第一步发生在30~275℃之间,这主要是由于样品中物理吸附的水和残留溶剂的蒸发所致[28].TGA曲线还显示了275~800℃范围为主要失重过程,这可能归因于碳层的燃烧.通过TGA分析,大致可确定在NiFe2O4@C-1,NiFe2O4@C-3和NiFe2O4@C-9中C的质量分数分别约为1.8%,5.5%,12.5%.为了进一步确定NiFe2O4@C复合材料中NiFe2O4和C的含量,通过ICP-OES测试了复合材料中各金属元素的含量,结果见表S2(本文支持信息).NiFe2O4@C-1,NiFe2O4@C-3,NiFe2O4@C-9复合材料中Ni的质量分数分别为24.58%,23.60%,21.76%,Fe的质量分数分别为47.04%,44.74%,41.03%,通过计算可以得到,复合材料中Ni和Fe的摩尔比接近1∶2,进一步可以计算出,在NiFe2O4@C-1,NiFe2O4@C-3,NiFe2O4@C-9复合材料中碳的质量分数分别为1.90%,5.81%,13.14%,这与TGA表征结果相接近.

Fig.4 TEM(A,C,E,G,I)and HRTEM(B,D,F,H)images of NiFe2O4(A,B),NiFe2O4@C-1(C,D),NiFe2O4@C-3(E,F,I),NiFe2O4@C-9(G,H),and EDS element mapping images of Ni(J),Fe(K),O(L),C(M)of NiFe2O4@C-3
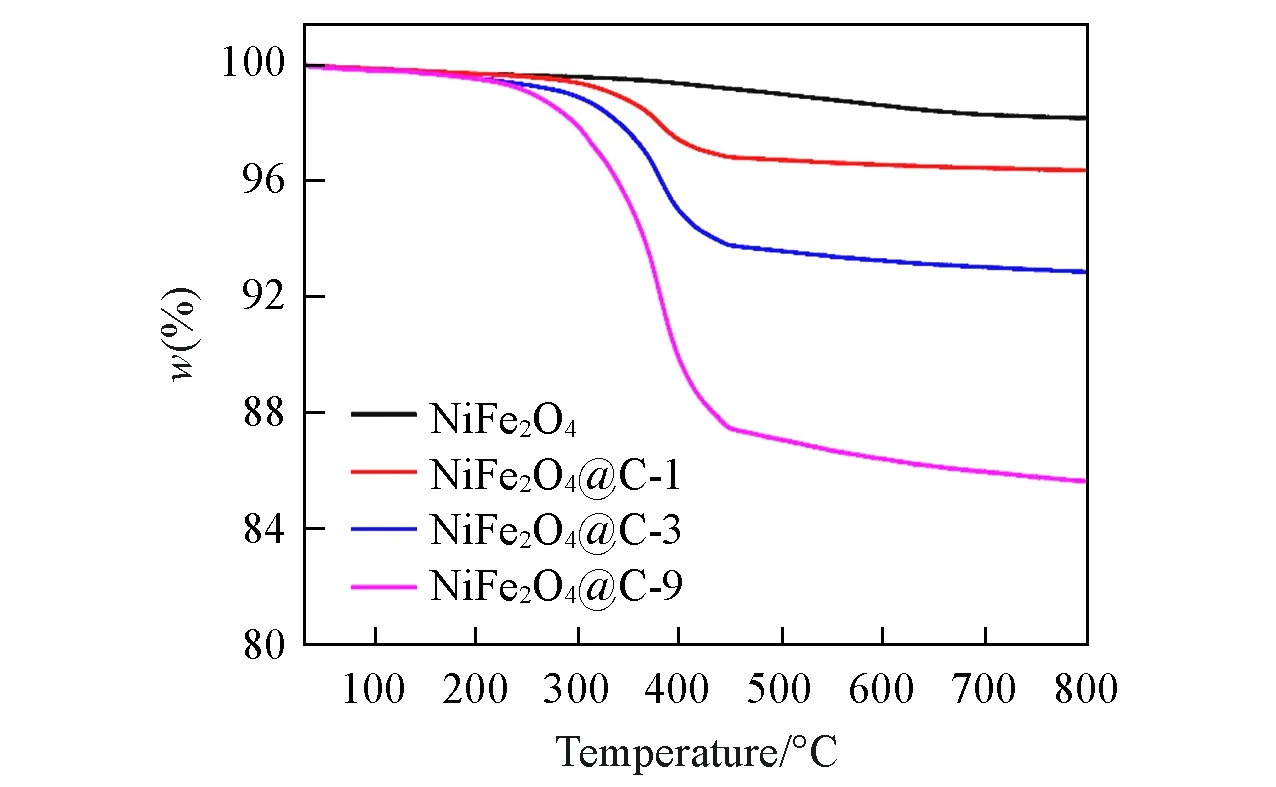
Fig.5 Thermogravimetric curve of the as-prepared NiFe2O4 and NiFe2O4@C composites
通过UV-Vis DRS光谱研究了NiFe2O4和NiFe2O4@C的吸光性能.从图6(A)可见,所有样品在200~800 nm范围内都有较好的吸光性能.相对于纯NiFe2O4,随着碳层厚度的增加,NiFe2O4@C样品在200~800 nm范围内的光吸收也随之增加.因此,NiFe2O4@C复合材料可能比纯的NiFe2O4具有更高的光催化活性.通过(ahv)2-hv关系曲线可以得到4个样品的能带间隙[图6(B)],通过计算得到NiFe2O4,NiFe2O4@C-1,NiFe2O4@C-3和NiFe2O4@C-9的禁带宽度分别为1.65,1.62,1.57和1.51 eV.可以看出,随着碳层厚度的增加,NiFe2O4@C的禁带宽度逐渐变窄,这有利于光催化性能的提升.
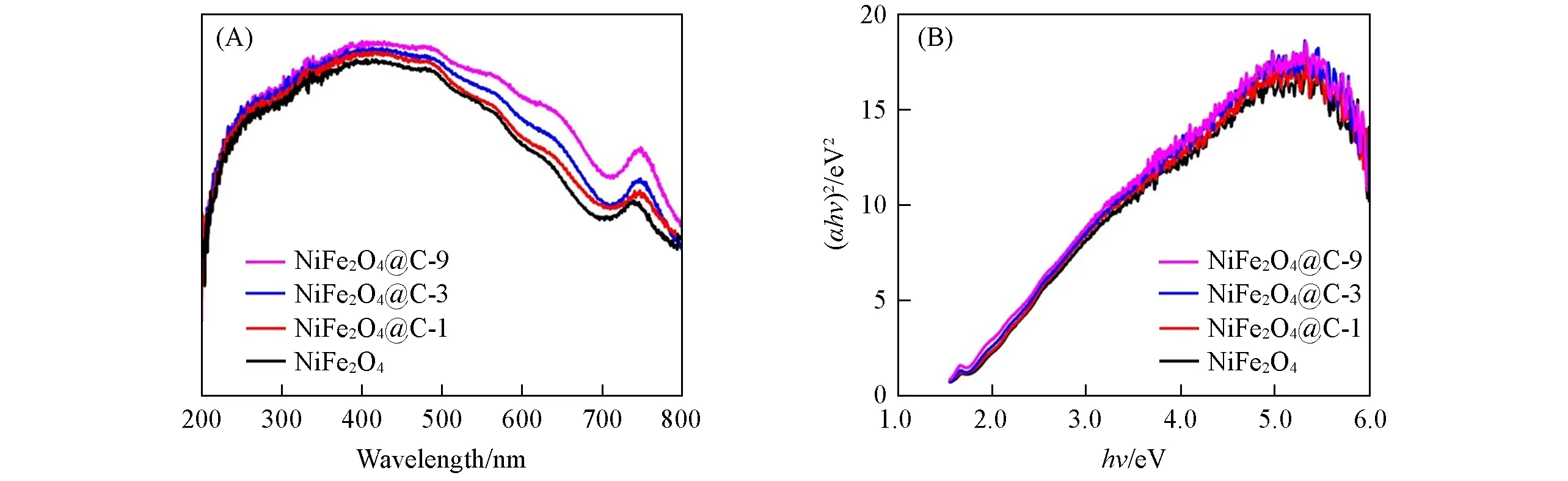
Fig.6 UV-Vis diffuse reflectance spectra of NiFe2O4 and NiFe2O4@C composites(A),plots of(ahv)2 vs.hv of the NiFe2O4 and NiFe2O4@C composites(B)
2.2 催化剂的光催化性能和稳定性
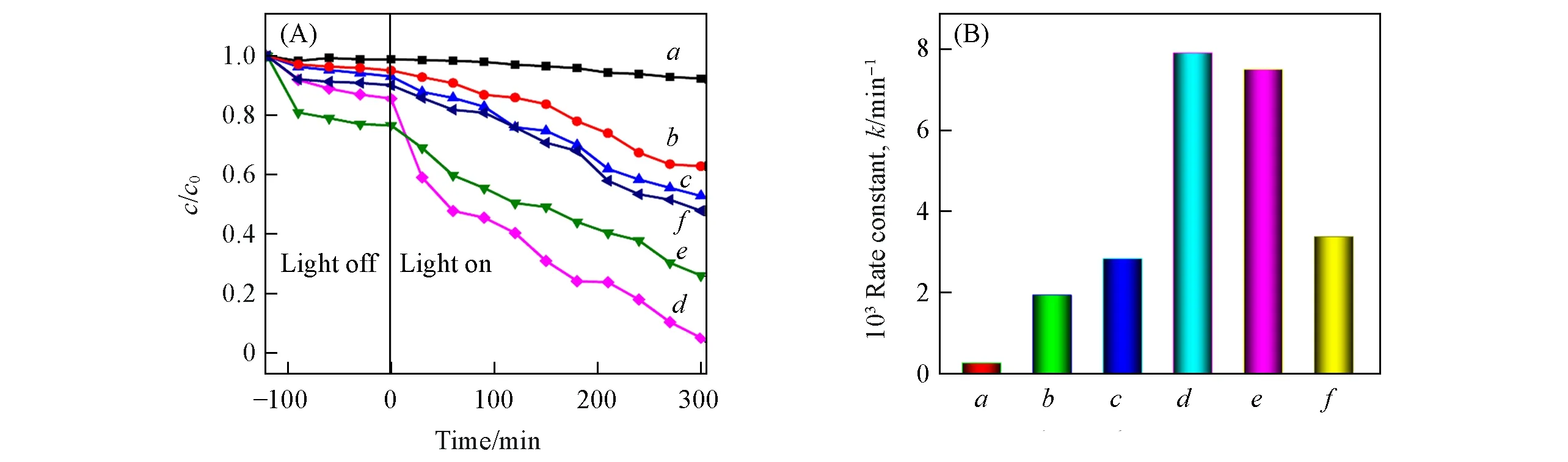
Fig.7 Photocatalytic degradation curves(A)and the reaction rate contrast(k)(B)for MB degradation by pure NiFe2O4,NiFe2O4@C composites and NiFe2O4+C mixturea.Blank;b.NiFe2O4;c.NiFe2O4@C-1;d.NiFe2O4@C-3;e.NiFe2O4@C-9;f.NiFe2O4+C.
为了评价NiFe2O4和NiFe2O4@C的光催化性能,研究了催化剂对MB的光催化降解性能.如图7(A)所示,在暗态下搅拌120 min,样品对MB的去除以吸附为主.NiFe2O4对MB的吸附率为4.9%,NiFe2O4@C-1,NiFe2O4@C-3和NiFe2O4@C-9对MB的吸附率分别为6.9%,14.4%和23.4%.可以看到,随着碳层厚度的增加,NiFe2O4@C对MB的吸附也随之增加.在光照下,样品对MB的去除以光催化降解为主.光照300 min后,没有任何催化剂时,MB的自降解率仅为7.6%.纯NiFe2O4对MB的光催化降解率为32.3%,NiFe2O4@C-1,NiFe2O4@C-3和NiFe2O4@C-9对MB的光催化降解效率分别为40.3%,80.7%和50.7%.很明显,碳包覆后的NiFe2O4,碳与NiFe2O4紧密接触形成异质结,导致复合材料光催化活性增强.当包裹的碳层厚度为5.5 nm时,NiFe2O4@C-3的光催化效率最高.然而,当碳层厚度继续增加到13.7 nm时,NiFe2O4@C-9的光催化活性反而不如NiFe2O4@C-3,这可能是因为,包覆太厚的碳层会阻止内部NiFe2O4对光的吸收,导致光生载流子数量减小[29],也可能是因为包覆太厚的碳层导致界面电阻增加,使载流子传输效率减小.为了验证NiFe2O4@C光催化活性的增强是由于碳与NiFe2O4紧密接触形成异质结构导致的,依据NiFe2O4@C-3中NiFe2O4和碳的比例,通过单纯物理混合制备了NiFe2O4+C混合物,并且测试了其光催化活性,结果如图7(A)所示.NiFe2O4+C物理混合物的光催化活性明显比NiFe2O4@C-3差,这是因为简单的物理混合很难形成紧密接触的异质结,导致光生载流子分离效率较差.这进一步证明通过表面水热碳层包覆半导体来构筑紧密接触的异质结,是提高光催化活性非常有效的方法.图7(B)为不同催化剂对MB光催化降解的一级动力学图,从图中可以明显看出,NiFe2O4表面包裹碳层确实可以有效提高其光催化活性.将相同质量的NiFe2O4@C-3分散到不同浓度(5,10,20,30,40 mg/L)的MB溶液中,来研究反应物初试浓度对光催化性能的影响(图S2,见本文支持信息),随着MB溶液初始浓度的增加,NiFe2O4@C-3对MB的降解率也随之减弱,这可能是因为高浓度的MB会覆盖催化剂表面的活性中心,同时也会抑制活性物种的产生[30].
催化剂的稳定性是评价其实际应用的重要指标,图S3(A)(见本文支持信息)展示了NiFe2O4@C-3催化剂5次循环光催化降解MB的情况.NiFe2O4@C-3重复使用5次以后,还能保持对MB 90.40%的去除率.与第一次(MB的去除率为95.09%)对比,光催化性能的降低非常小,这说明所制NiFe2O4@C-3有较好的可持续使用性.图S3(B)展示了NiFe2O4@C-3使用前后的XRD谱图,催化剂反应前后的XRD谱图基本一致,这说明NiFe2O4@C-3具有较好的稳定性.图S3(C)对比了NiFe2O4@C-3分散在水中,有无外加磁铁的分离情况.在外加磁铁的作用下,NiFe2O4@C-3很容易从水中分离,说明NiFe2O4@C非常容易回收利用.总之,NiFe2O4@C复合材料展现了较高的光催化活性、较好的可持续使用性和稳定性,易分离、可回收,在光催化降解水中污染物方面有很大的应用潜力.
2.3 光催化性能的增强机制
通过PL光谱、瞬态光电流和EIS谱探究了表面水热碳层对NiFe2O4光生载流子分离和转移以及光催化活性的影响机制.图8(A)为不同样品的荧光光谱图,对于半导体光催化剂,光生载流子的迁移和复合与荧光光谱强度有关,荧光光谱越弱,表明光生载流子分离效率越好[31].从图8(A)可见,NiFe2O4在400~600 nm范围内有宽且较强的发射峰,碳包覆NiFe2O4以后,荧光光谱的强度均明显减弱,这说明碳包覆可以增强NiFe2O4光生载流子的分离效率.其中,NiFe2O4@C-3的荧光峰强度最小,表明其光生载流子复合几率最小.图8(B)展示了不同样品的瞬态光电流响应图.可见,在光照下,纯的NiFe2O4几乎没有光电流响应,而NiFe2O4@C样品都有较强的光电流,这进一步说明碳包覆可有效促进NiFe2O4光生载流子的分离.其中,NiFe2O4@C-3具有最高的光电流响应,表明其光生载流子的分离效率最高,这一结果与荧光表征相一致.值得注意的是,相对于NiFe2O4@C-3,NiFe2O4@C-9的光电流轻微减弱,这可能是因为包覆太厚的碳层阻碍了内部NiFe2O4对光的吸收,导致光生载流子数量减小,这也进一步解释了NiFe2O4@C-9光催化活性降低的原因.通过电化学阻抗谱研究了载流子的转移情况,如图8(C)所示,碳包覆NiFe2O4以后,Nyquist曲线圆弧明显减小,越小的圆弧半径表明界面接触电阻越小,光生载流子迁移效率越高[32].NiFe2O4@C-3的Nyquist曲线圆弧半径最小,表明其界面接触电阻最小,光生载流子迁移效率最高.相对于NiFe2O4@C-3,NiFe2O4@C-9的Nyquist曲线圆弧半径变大,这可能是因为包覆太厚的碳层使界面电阻增加,光生载流子迁移效率减小,使其光催化活性降低.总之,通过PL、瞬态光电流和EIS表征证明,碳包覆NiFe2O4有效促进了光生载流子的分离和传输效率,导致光催化活性明显增强.但是,包覆太厚的碳层会阻止内部NiFe2O4对光的吸收,导致光生载流子数量减小,此外,太厚的碳层也导致界面电阻增加,使载流子的传输效率减小.
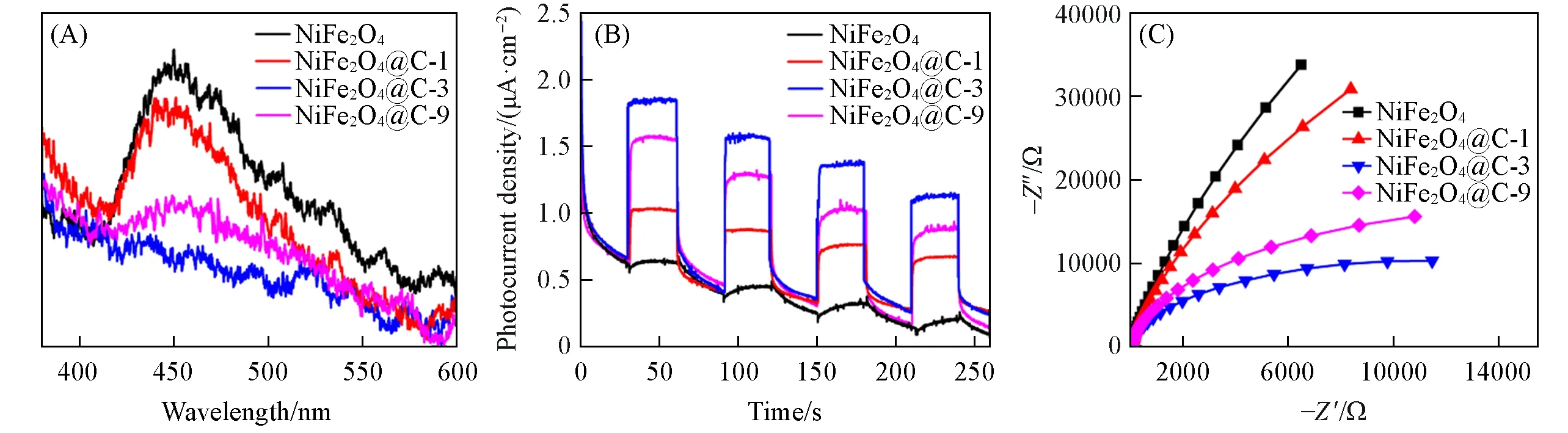
Fig.8 PL spectra(excited at 325 nm)(A),photocurrent response(B)and Nyquist plots(C)of pure NiFe2O4 and NiFe2O4@C composites
2.4 光催化机理
为了深入研究光催化机理,通过自由基捕获实验来探究NiFe2O4@C-3在光催化反应中的活性物种.在其它实验条件不变的情况下,向MB溶液中分别加入1 mmol的TBA,EDTA-2Na,AgNO3和BQ来捕获羟基自由基(·OH)、空穴(h+)、电子(e-)和超氧自由基(·O2-)[33].从图9可见,不加任何猝灭剂时,MB的去除率高达95.09%,加入TBA和EDTA-2Na后,MB的降解效率轻微降低,依然能保持到83.23%和71.65%.而加入AgNO3和BQ后,MB的降解受到严重抑制,去除率分别降低到32.17%和22.12%.这说明在光催化反应过程中,e-和·O2-是主要的活性物种.
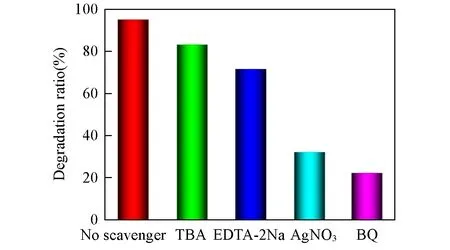
Fig.9 Trapping experiments of active species during the photocatalytic degradation of MB over NiFe2O4@C-3
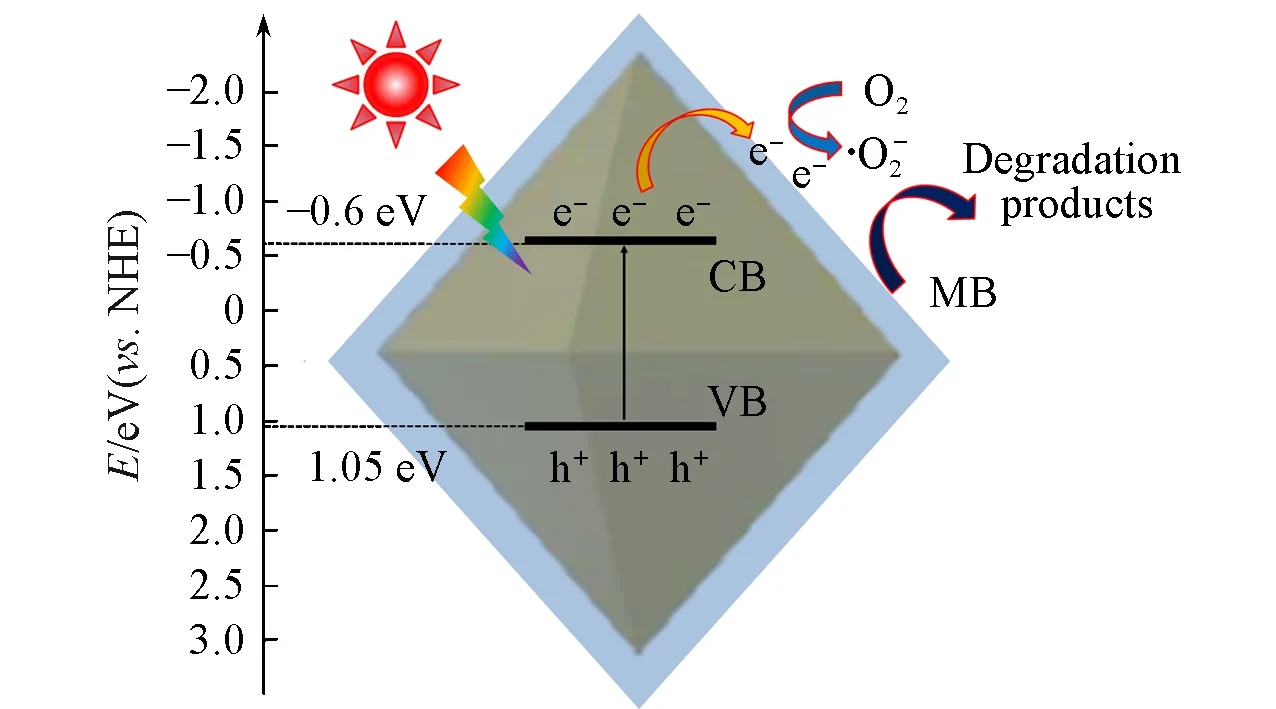
Fig.10 Possible mechanism of photocatalytic degradation MB by NiFe2O4@C-3 composite under light irradiation
基于以上分析,推测NiFe2O4@C-3复合材料可能的光催化反应机理如图10所示.在光照下,半导体NiFe2O4被激发产生e-和h+,由于碳材料具有良好的电子传输性能,产生的e-很容易通过碳层转移到催化剂的表面,h+则留在NiFe2O4的价带,这导致电子和空穴有效分离.转移到催化剂表面的e-与吸附的O2反应产生·O2-,强氧化性的·O2-将MB氧化降解.因此,在光催化过程中,引入e-与·O2-捕获剂会严重阻碍MB的降解效率.而由于碳层的包覆降低了NiFe2O4与MB的接触几率,阻碍了NiFe2O4价带上的h+对MB的降解.同时,由于NiFe2O4的价带位置(1.05 eV)比OH-/·OH的氧化还原电位(2.4 eV)更负,留在NiFe2O4价带上的h+不能氧化OH-产生·OH[34].因此,在光催化反应过程中·OH和h+不是主要的活性物种.
3 结论
采用葡萄糖水热碳化法合成了磁性NiFe2O4@C核壳八面体.通过调控葡萄糖的含量控制NiFe2O4表面包覆的碳层厚度在3.7~13.7 nm之间.研究了表面水热碳层对NiFe2O4光催化降解MB性能和稳定性的影响.结果表明,NiFe2O4核和碳壳之间形成了紧密接触的异质结,有效地促进了光生载流子的分离和传输效率.表面水热碳层厚度为5.5 nm时,NiFe2O4@C-3展现了最佳的光催化活性.表面水热碳层包覆既能阻止磁性NiFe2O4纳米粒子的团聚,又能利用其较好的电子传输性能促使NiFe2O4光生电子的迁移,提高其光催化活性,研究结果有望为高活性半导体核/壳结构光催化剂的设计提供新的思路.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20220472.
猜你喜欢
陶瓷学报(2021年3期)2021-07-22 01:05:06
原子与分子物理学报(2021年2期)2021-03-29 07:30:58
新世纪智能(数学备考)(2019年9期)2019-10-16 11:44:58
物理学报(2019年1期)2019-01-25 09:53:52
物理学进展(2017年1期)2017-02-23 01:35:44
西安工程大学学报(2016年6期)2017-01-15 14:08:22
载人航天(2016年3期)2016-06-04 06:08:42
湖北师范大学学报(自然科学版)(2015年1期)2016-01-10 08:41:29
陶瓷学报(2015年4期)2015-12-17 12:45:02
大学化学(2015年5期)2015-09-18 08:43:48
