
低温等离子体协同Mn基催化剂降解氯苯研究
2022-11-13 07:31石秀娟梁文俊尹国彬王金柱
化工学报 2022年10期
石秀娟,梁文俊,尹国彬,王金柱
(北京工业大学区域大气复合污染防治北京市重点实验室,北京 100124)
引 言
挥发性有机化合物(volatile organic compounds,VOCs)是大气环境中主要污染物之一,具有易挥发、来源广、成分复杂等特点。它是形成臭氧(O3)和细颗粒物(PM2.5)污染的重要前体,因对人类健康和环境存在严重威胁而受到广泛关注[1-3]。其中,含氯挥发性有机物(chlorinated volatile organic compounds,CVOCs)具有高毒性、高稳定性且难生物降解[4-5]等特性,因而在《斯哥德尔持久性有机污染物公约》中被列入有限控制有机污染物[6]。近年来,VOCs 末端控制技术的研究主要集中在吸附法[7]、催化氧化法[8]、光催化法[9]和燃烧技术[10-11]等,但传统的控制技术对低浓度VOCs 的治理存在一定的缺陷[12-13]。而低温等离子体(non-thermal plasma,NTP)技术因其可在常温常压下工作、操作简单,可同时处理多种污染物,以及随时启停等优点而被广泛应用于VOCs 的降解,尤其是中低浓度VOCs 的降解[14]。但单独采用NTP 技术降解VOCs 也存在一定的缺点,如降解效率低、能量利用率低以及生成有毒有害副产物等[15-17]。
近年来的研究表明,在反应系统中引入催化剂可有效提高VOCs 降解率、能量利用率以及降低有害副产物的产生[18]等。在NTP 协同催化反应系统中,催化剂的选择至关重要,因此国内外学者对此进行了大量的探索研究,研究发现不同活性组分催化剂对VOCs 降解效率、副产物生成以及相关机理略有不同;相同金属催化剂,其制备方式、活性组分氧化物价态及与NTP 结合方式的不同也会对VOCs降解活性产生影响[19]。过渡金属Mn 因价格低廉且具有较好的低温活性而被广泛应用,同时MnOx催化剂因多晶型、多价性和高储氧能力等优势而引入NTP 协同催化反应体系中开展研究[20-21]。 Mn 基催化剂分散性较好,反应系统中产生的O3能够在催化剂表面分解为活性氧,可进一步提高反应系统中VOCs 的降解性能[22]。Liu 等[23]在DBD 反应器中引入MnOx催化剂,氯苯降解效率、COx选择性以及O3抑制均得到提升,低电压条件下效果更为明显;在输入能量密度为1350 J∕L 时,氯苯降解效率达到96.3%,反应系统中O3在MnOx表面分解形成氧原子以氧化氯苯,从而提升了氯苯降解效率。因此,NTP 协同Mn基催化剂反应系统中,活性组分在载体表面的分散性至关重要。研究表明[24],对于Mn 基催化剂,不同的前体制备的催化剂会影响Mn 元素在催化剂表面的分散性和催化活性。张硕等[25]研究表明,以乙酸锰为前体制备的催化剂对甲苯的氧化能力更强,该催化剂具有较大的比表面积,有利于活性组分在载体表面的高度分散,有利于甲苯的降解和O3的分解;与单独DDBD 相比较,其COx转化率提升39.2%,O3降低33.4%。Wang 等[26]探究不同前体制备的Mn基催化剂对邻二甲苯降解性能的影响,得出相似的实验结果。而氯苯作为一种典型的CVOCs,广泛应用于涂料、印刷、石化、农药制造和氯碱工业等领域,是二英等剧毒物质的重要前体物[5,23,27]。氯苯在NTP 协同催化反应体系中会发生C—Cl 键的断裂和解离,产生的含氯中间产物可能会造成催化剂的失活[28]。现阶段,NTP 协同Mn 基催化剂降解氯苯的研究不少,但催化剂制备前体对氯苯降解性能以及臭氧生成情况的研究尚不系统,其研究结果是否与甲苯以及邻二甲苯等污染物一致有待探究。
本文分别采用乙酸锰和硝酸锰为前体,γ-Al2O3为载体制备催化剂,并与NTP 协同降解氯苯,对氯苯降解效率、能量效率以及臭氧生成情况等开展研究。利用N2吸附-脱附、XRD 以及FT-IR 等测试方法对反应前后催化剂进行表征分析,对不同前体催化剂对氯苯降解性能以及臭氧生成差异进行对比分析,同时分析了反应过程中NTP 对催化剂结构、性能的影响。
1 实验材料和方法
1.1 催化剂制备
实验所用催化剂均采用浸渍法制备,分别以硝酸锰和乙酸锰为前体,γ-Al2O3小球作为载体制备催化剂,即配制一定浓度的硝酸锰和乙酸锰溶液,称取一定质量的γ-Al2O3小球分别加入配制好的前体溶液中,超声浸渍2 h后再避光浸渍3 h,然后放入水浴锅进行加热处理3 h,将负载催化剂放入烘箱中在110℃下干燥3 h,在500℃下焙烧3 h,得到MnOx(MN)∕γ-Al2O3和MnOx(MA)∕γ-Al2O3催化剂。
1.2 催化剂表征
催化剂的比表面积和孔结构通过Micromeritics ASAP 2050 自动物理吸附仪进行分析,分别通过Brunauer-Emmet-Teller(BET) 和 Barrett-Joyner-Halenda(BJH)方法计算。采用日立SU 8220(日本)扫描电镜(SEM)对催化剂表面形貌进行观察和分析。采用D8 Advance 型X 射线衍射仪(德国Bruker∕AXS公司)进行X 射线衍射(XRD)测定。对催化剂进行晶体结构分析,采用Cu Kα放射源,扫描速率5(°)∕min,扫描步长0.02°,扫描范围2θ=5°~90°。X 射线光电子能谱(XPS)可对催化剂表面元素价态和含量进行分析。使用ESCALAB 250XI(Thermo,American)和Al Kα辐射(hν=1486.8 eV)对催化剂进行XPS 分析。所有结合能(BE)均通过284.8 eV 的C 1s 结合能进行校正。采用美国Thermo Fisher Scientific 公司Nicolet型红外光谱仪对催化剂组成进行分析,被测样品粉末与干燥后KBr 混合后压片制样,图谱采集范围为400~4000 cm-1,步长0.4 cm-1,扫描次数16 次。采用离子色谱仪(Metrohm,883 Basic IC plus)对氯离子浓度进行测定,被测离子通过大气采样仪对反应尾气吸收,吸收液为0.01 mol∕L NaOH 溶液,以1 L∕min 的速率采气30 min。采用气相色谱-质谱联用仪(Thermo,Trace DSQ,USA)定性分析反应过程中产生的中间产物。
1.3 实验装置及条件
本实验在常温常压下进行。实验装置如图1所示,主要包括配气单元、等离子体催化反应单元、高压电源和气体检测四部分。背景气体采用氮气和氧气配气,总进气流量控制为5 L∕min,其中氮气分为两路:一路经流量计后鼓入氯苯发生瓶,恒温条件下水浴加热确保氯苯分子挥发进入混气瓶,另一路气体经流量计后直接进入混气瓶,氧气经流量计后直接进入混气瓶,三路气体进入混气瓶混合趋于稳定后进入等离子体催化反应器。反应后气体进入气相色谱仪(6890N, Agilent)中分析,反应过程中产生的臭氧采用臭氧分析仪(106-M,2B Technology)测定,反应器表观温度采用热成像仪(Testo 885-2)测定。低温等离子体催化反应器采用管线式结构,反应器材质为石英玻璃(外径32 mm, 内径29 mm),以钨丝作高压电极,铝箔作接地极,放电区长度为100 mm,实验前在放电区中间填充50 mm 催化剂。实验采用变频变压交流电源,调压范围0~100 kV,调频范围0~1000 Hz。
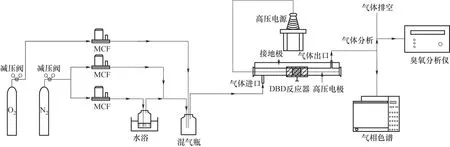
图1 实验装置Fig.1 Schematic diagram of experimental system
实验过程中采用连续进气方式操作,即含有污染物的气流连续通过装有催化剂的反应管,在催化剂达到吸附饱和后继续通入气流,污染物浓度在30 min 内保持相对稳定即作为污染物进口浓度,实验过程中氯苯初始浓度控制在500 mg∕m3。
1.4 评价指标
氯苯净化效果以η(%)作为评价指标,具体表达式如式(1)所示;等离子体反应系统的放电能量水平高低以输入能量密度SED(J∕L)作为评价指标,具体表达式如式(2)所示;反应系统中氯苯降解能耗评价以能量效率EE[g∕(kW·h)]作为评价指标,具体表达式如式(3)所示。
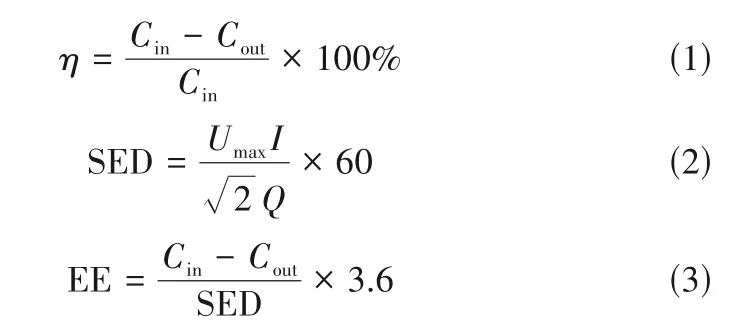
式中,Cin、Cout分别为氯苯进出口浓度, mg∕m;Umax为峰值电压,kV;I为电流,mA;Q为气体流量,L∕min。
2 实验结果与分析
2.1 低温等离子体协同催化降解氯苯研究
2.1.1 不同电压条件下催化剂对SED 的影响 低温等离子体协同催化反应系统降解污染物过程中,放电电压以及填料的引入均对反应系统内SED 产生影响。
图2为不同电压条件下催化剂引入对反应系统SED 变化的影响。从图中可以发现,不同催化剂反应系统内SED 随放电电压升高而变大;填充不同种类催化剂,其SED也不同。相同电压条件下,SED顺序 为:NTP+MnOx(MN)∕γ-Al2O3≈NTP+MnOx(MA)∕γ-Al2O3>NTP+γ-Al2O3>NTP。与单独NTP 相比,反应系统内引入填料后其SED 增大,分析其原因,可能是在NTP 协同催化反应系统内,填充物质在电场作用下会产生较强的局部放电现象,且填料的引入会减小放电间隙,从而增大反应系统的SED[29]。而负载活性组分填料反应系统SED 高于未负载活性组分填料的反应系统,原因归结为填料物质的介电常数ξ不同,ξ(γ-Al2O3)为9.34~11.54,而ξ(MnOx)约为104,使 得ξ[MnOx(MN∕MA)∕γ -Al2O3] 大 于ξ(γ -Al2O3),介电常数小的物质在催化剂颗粒之间不能形成有效的微放电,从而导致反应系统的SED降低[30]。
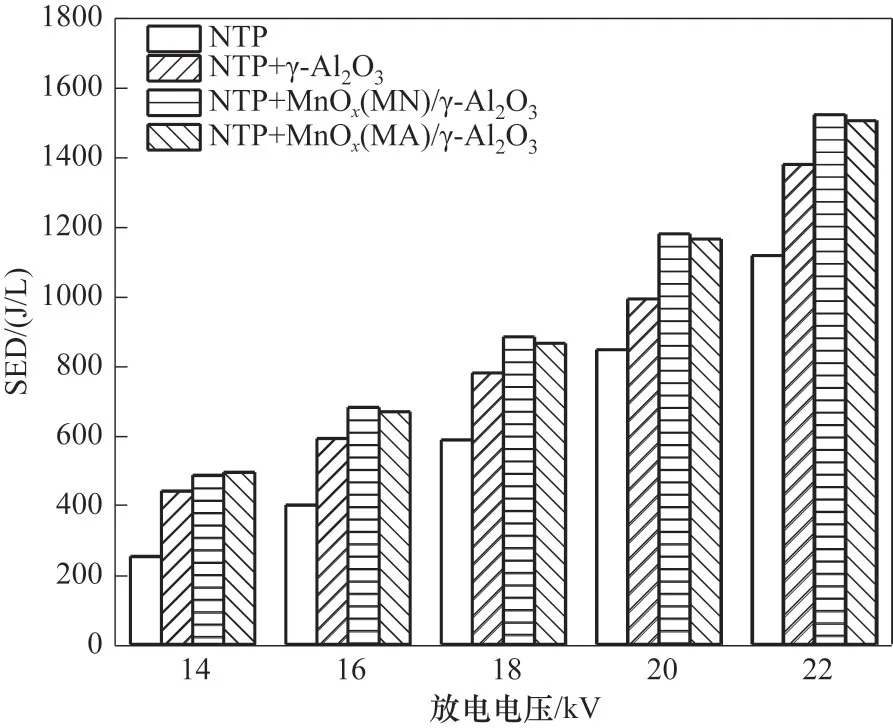
图2 不同放电电压条件下催化剂对SED的影响Fig.2 Effect of catalyst on SED under different discharge voltage conditions
2.1.2 不同电压条件下催化剂对氯苯降解效率和能量效率的影响 采用NTP 在空气气氛下进行氯苯降解研究,考察了放电电压和不同填料对氯苯降解效率及EE 的影响,结果如图3 所示。由图3 可知,催化剂的引入明显提高了反应系统内氯苯的降解效率和EE。在放电电压为14 kV 时,单独NTP 降解氯苯的去除效率为33.9%,引入γ-Al2O3后氯苯降解效率提升至67.4%,负载活性组分MnOx(MN∕MA)∕γ-Al2O3后氯苯降解效率>75%;而不同反应系统[NTP、NTP+γ-Al2O3、NTP+MnOx(MN)∕γ-Al2O3、NTP+MnOx(MA)∕γ-Al2O3]的EE 分别为2.4、2.8、2.9、3.0 g∕(kW·h),催化剂的引入提高了反应系统的EE。相同条件下,不同填料引入后氯苯降解效率顺序为:NTP+MnOx(MA)∕γ-Al2O3>NTP+MnOx(MN)∕γ-Al2O3>NTP+γ-Al2O3>NTP。产生该现象的原因可能是反应器中填料的引入对氯苯具有一定的吸附作用,从而延长了氯苯在反应器内的停留时间并提高了系统内的介电常数,从而提高了反应系统内氯苯的降解效率;同时,填料的引入也提高了系统内的放电强度,其促进高能活性氧原子的电离形成和向更高能量水平的转移[31],提升了系统内温度和能量水平,促进臭氧在催化剂表面的分解,分解产生的活性氧原子参与到氯苯降解反应中,从而提高反应系统内氯苯降解效率和EE。
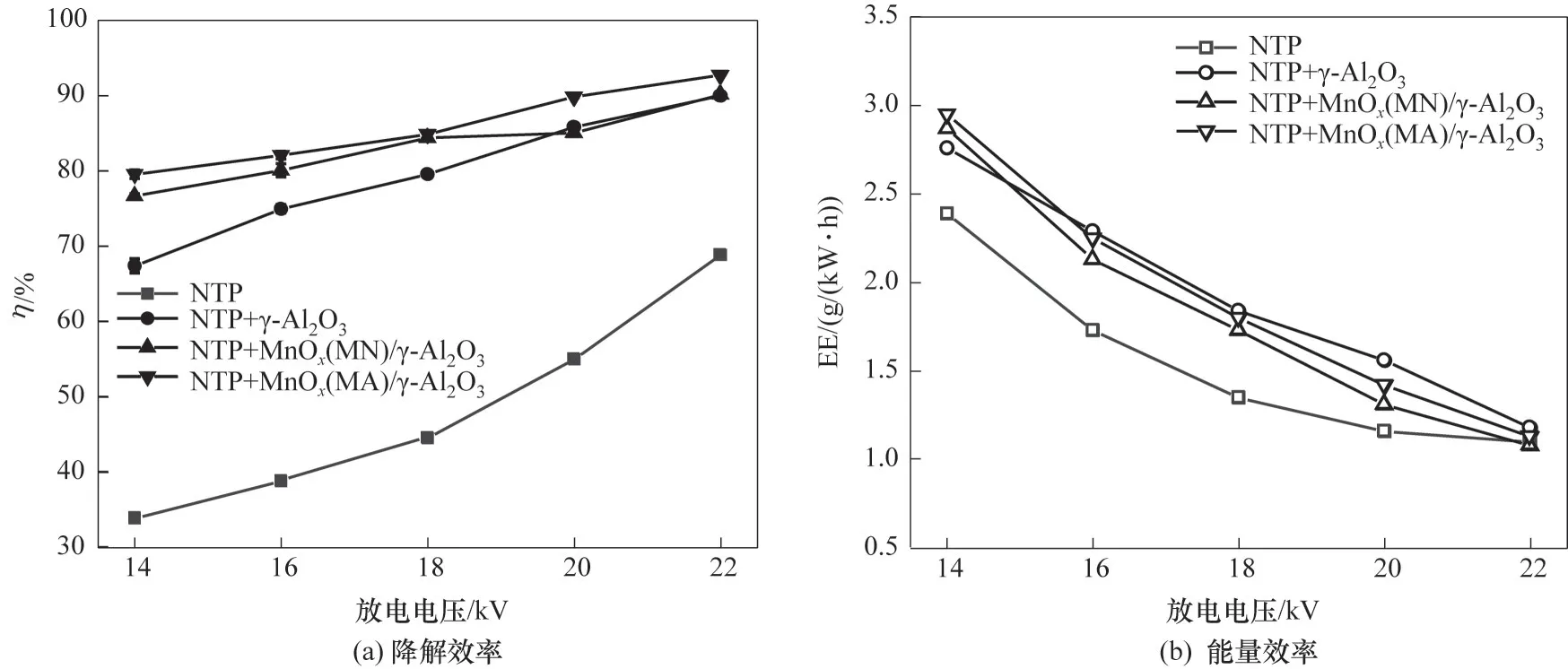
图3 不同放电电压条件下催化剂对氯苯降解效率和能量效率的影响Fig.3 Effect of catalyst on degradation efficiency and EE of CB under different discharge voltage
相同条件下MnOx(MA)∕γ-Al2O3对氯苯取得更佳降解效率,且随放电电压从14 kV 提高到22 kV,氯苯的降解效率从79.6%提高到92.8%,究其原因可能是放电电压增加,导致反应系统产生更多的活性粒子与高能粒子,与氯苯的碰撞概率增加,从而提高了氯苯的降解效率;在NTP 协同Mn 基催化剂反应系统中,背景气体中氧气、放电过程中产生的臭氧、电子和自由基在催化剂表面通过Mn 价态之间相互转化,并参与到VOCs 降解过程中,大大提高了氯苯的降解效率[32]。通过催化剂比表面积SBET、XRD和XPS 表征可以发现,乙酸锰为前体制备的Mn 基催化剂具有更大的比表面积,活性组分在催化剂表面分散性更好,臭氧更容易在催化剂表面发生分解,生成氧化能力更强的活性氧原子与氯苯发生反应,从而提高氯苯的降解效率。在低电压(14 kV)条件下,NTP+MnOx(MA)∕γ-Al2O3反应系统中氯苯降解效率比单独NTP 系统高45.7%,而在高电压(22 kV)条件下这一差值降至23.9%,说明在较低电压条件下催化剂引入对氯苯降解效率的提升更为明显,这主要是由于高电压条件虽然为反应系统提供了更多的活性粒子,增加氯苯与活性粒子的碰撞概率,但反应系统内一部分能量转化为光能和热能,在22 kV时反应器表观温度达到150℃,会产生火花放电,从而消耗大量的能量,导致反应系统内氯苯降解效率提升幅度较小,因此较高电压条件下催化剂的加入对氯苯降解效率的提升作用较低[33]。
2.2 催化剂的表征
2.2.1 N2吸附-脱附曲线和孔径分布 不同催化剂协同NTP 降解氯苯反应前后的N2吸附-脱附曲线及孔径分布如图4 所示。从图中可以发现,不同催化剂反应前后的吸附等温线确定为Ⅳ型等温线,在相对压力(P/P0)处于0.4~1.0 时呈现H3 型滞后环,这是介孔材料的典型特征[34-35],且各种催化剂存在较小的介孔。反应前后催化剂孔径分布的峰位集中在2.5~10 nm,最可几孔径在6~7 nm 之间,也说明催化剂中存在较小的介孔。同时放电前后催化剂吸脱附曲线及孔径分布情况说明,放电过程并未对催化剂的孔道结构产生破坏。表1为等离子体反应前后不同催化剂的比表面积SBET、总孔容Vp和平均孔径dp数据。由表1 可知,γ-Al2O3的SBET为257 m2∕g,而引入活性组分后SBET减小,说明大量活性组分的负载使催化剂表面褶皱或孔道被填充,从而导致其SBET减小。结合氯苯降解活性数据可知,氯苯降解效率与催化剂的SBET之间并不是呈线性关系。虽然γ-Al2O3的SBET最高,但污染物降解效率并不是最好的(图3),这说明较大的SBET虽然有利于污染物物理吸附[36],但不是影响催化活性的唯一因素。而经等离子体处理后,催化剂的SBET和Vp有所增大,dp有所降低,但变化幅度均不大,该实验结果与文献[37-38]实验结果基本一致。
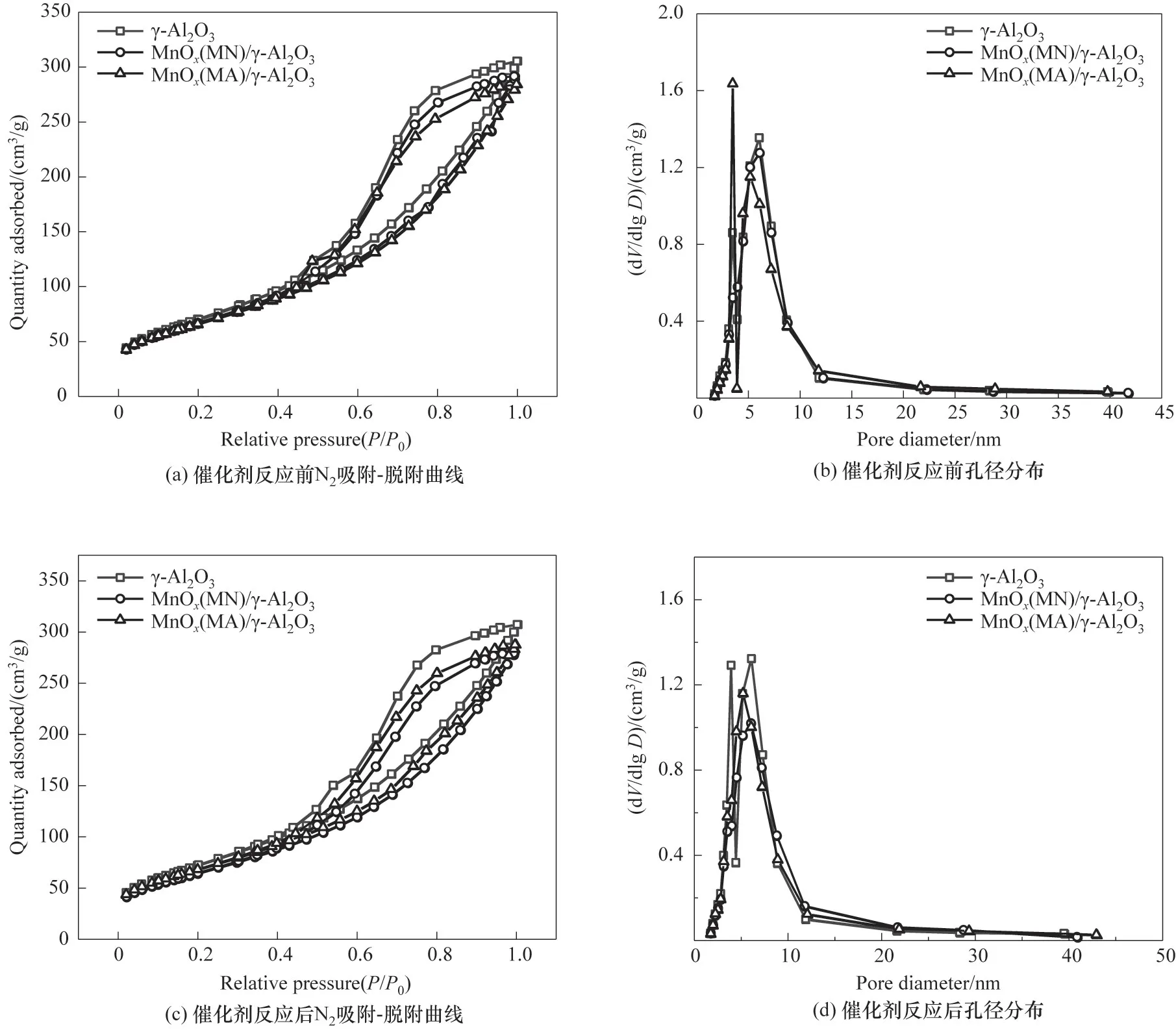
图4 不同催化剂反应前后的N2吸附-脱附曲线和孔径分布Fig.4 N2 adsorption-desorption curves and pore size distribution before and after reaction with different catalysts
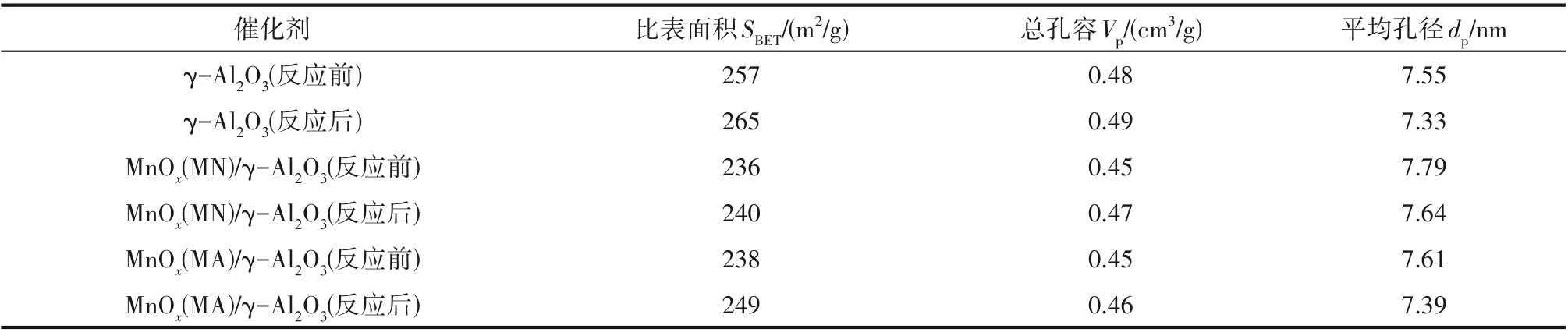
表1 不同催化剂反应前后的比表面积、孔容和孔径Table 1 Specific surface area,pore volume and pore diameter of different catalysts before and after reaction
2.2.2 催化剂SEM 表征 为考察不同前体活性组分在γ-Al2O3小球表面的负载情况以及不同催化剂的表面形貌,选取γ-Al2O3小球原样、MnOx(MN)∕γ-Al2O3和MnOx(MA)∕γ-Al2O3进行了扫描电镜分析,具体结果如图5所示。
从图5 可以看出,未负载活性组分的γ-Al2O3小球载体表面具有较多的空腔和较多较大的凹陷处,不同前体的Mn 活性组分的加入,使得MnOx(MN∕MA)∕γ-Al2O3表面变得致密,因此催化剂SBET减小,dp增大,且催化剂表面的凹陷被活性组分占据而呈现凸起,从而增大了催化剂表面反应活性中心数量,提高了氯苯的降解效率;与MnOx(MN)∕γ-Al2O3相比较,MnOx(MA)∕γ-Al2O3的SBET相对较大,活性组分在催化剂表面负载更为均匀、致密。

图5 不同催化剂扫描电镜照片Fig.5 SEM images of different catalysts
2.2.3 XRD 分析 为进一步研究不同催化剂协同NTP 降解氯苯反应前后催化剂的晶相变化,对反应前后催化剂进行XRD分析,结果如图6所示。
从图6(a)中可以发现,在2θ=37.538°、45.666°、66.600°处观察到具有立方尖晶石结构的γ-Al2O3(PDF#50-0741)衍射峰。图6(a)中,对于MnOx(MN)∕γ-Al2O3,除γ-Al2O3的衍射峰外,在2θ=32.920°处出现微弱的新峰,对比PDF#24-0508 可知,此峰归属于Mn2O3。对于MnOx(MA)∕γ-Al2O3,未发现MnOx的衍射峰,而分析催化剂FT-IR 发现,催化剂在570~580 cm-1处出现的宽峰为MnO6八面体骨架中Mn-O振动峰,从而说明MnOx可能高度分散于载体γ-Al2O3表面,因此乙酸锰在载体表面分散性更好。
由放电后催化剂的XRD 谱图[图6(b)]发现,放电前后γ-Al2O3和MnOx(MA)∕γ-Al2O3中各组分衍射峰未发生明显变化,而放电后MnOx(MN)∕γ-Al2O3中未发现MnOx衍射峰,可能是放电过程中产生的物质对MnOx进行掩盖,从而不能观察到Mn2O3衍射峰。同时XRD 测试结果总体说明,放电并没有改变催化剂的晶型结构。
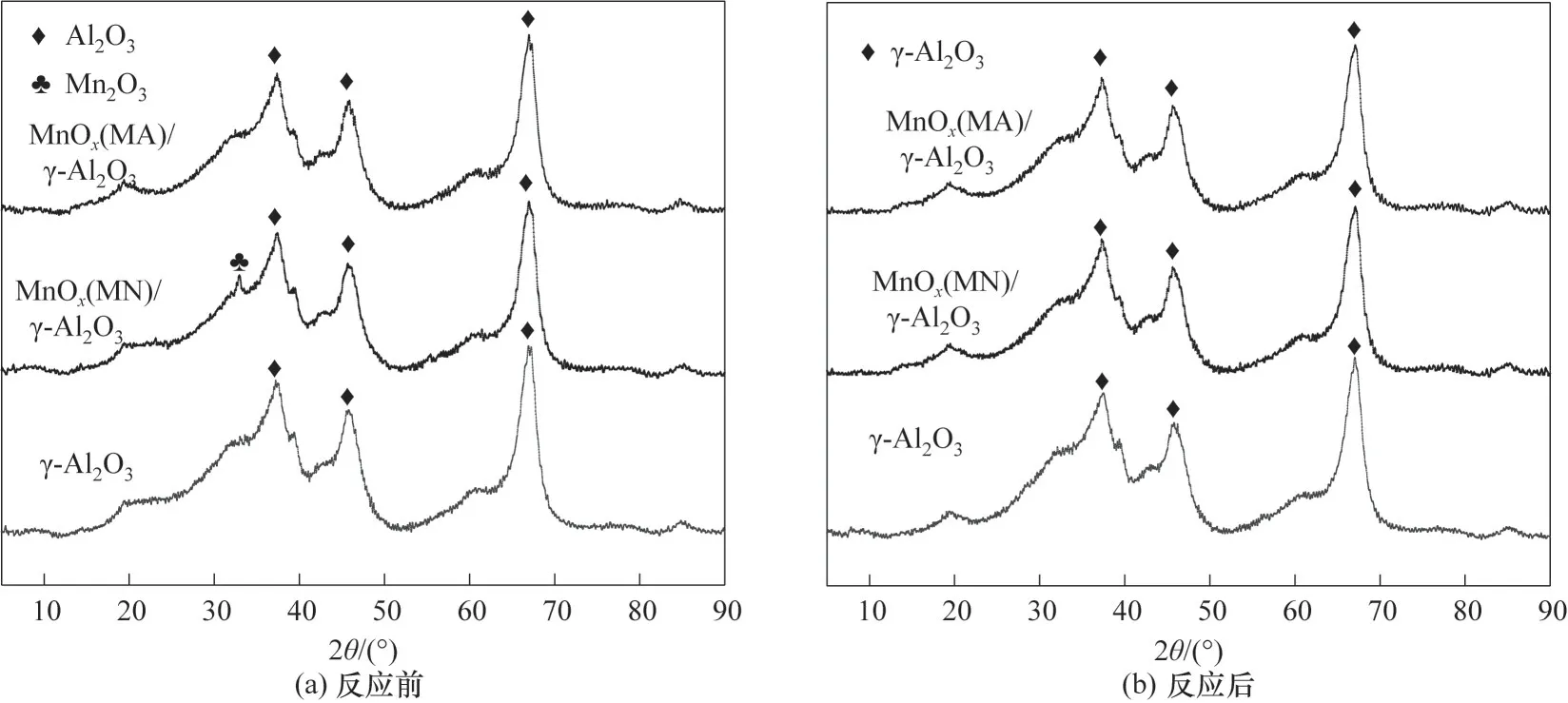
图6 不同催化剂反应前后的XRD谱图Fig.6 XRD patterns of different catalysts before and after reaction
2.2.4 FT-IR 分析 使用FT-IR 对催化剂的结构和化学组成进行分析确认,结果见图7。如图所示,反应前后所有催化剂在3400 和1600 cm-1附近出现吸收峰,分别归属于O—H 伸缩振动和O—H 弯曲振动,表明催化剂表面存在OH 基团[39],而文献[25]表明,OH 基团对VOCs 的降解过程起重要作用。反应前后催化剂在2300 cm-1处出现的微弱吸收峰是由CO2反伸缩振动引起的。对比催化剂反应前后1400 cm-1附近的吸收峰可以发现,反应后催化剂的振动强度增大,可能是反应过程中中间产物与催化剂结合生成某些碳酸盐物质。在570~580 cm-1处出现的宽峰为MnO6八面体骨架中Mn-O 振动峰,这表明Mn-O 键的形成[40-41]。通过分析红外结果可以发现:MnOx在载体表面成功负载,且反应过程中中间产物与催化剂结合可能生成的某些碳酸盐类物质会附着在催化剂表面,从而对后续实验产生影响。
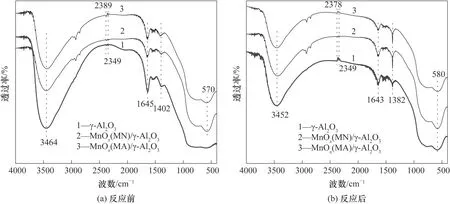
图7 不同催化剂反应前后的FT-IR谱图Fig.7 FT-IR spectra of different catalysts before and after reaction
2.2.5 XPS 分析 为研究催化剂表面Mn 和O 组分的化合价价态以及含量,对不同前体制备的新鲜Mn基催化剂进行了XPS 表征分析,结果如图8 和表2所示。图8(a)为Mn 2p3∕2的XPS 谱图。在不同前体制备的新鲜Mn 基催化剂样品中都可以观测到不对称Mn 2p3∕2峰,经拟合后分解为640、641.7、643.7 eV左右的三个峰,分别归属于Mn2+、Mn3+、Mn4+,说明制备的新鲜Mn 基催化剂中Mn 元素以不同的价态存在。O 1s 的XPS 谱图如图8(b)所示,每个催化剂的不对称的O 1s XPS 谱图可分解为三个组分,其中结合能为529.2 (529.3) eV 的组分归属于表面晶格氧(Olatt)物种,结合能为530.6 (530.8) eV 的组分归属于表面吸附氧(Oads)物种,结合能为532.2 (532.3) eV 的组分归属于表面吸附水或碳酸盐物种[32]。有研究表明[26,40],在等离子体反应系统中,Mn4+与晶格氧(Olatt)所占比例越高,越有利于VOCs 的去除,Mn4+可有效分解反应系统内O3生成活性氧原子。本实验MnOx(MN)∕γ-Al2O3和MnOx(MA)∕γ-Al2O3催化剂中,Mn4+∕Mn3+的值分别为0.77 和0.88,Olatt∕Oads的值分别为2.56 和3.91(表2)。结果表明,MnOx(MA)∕γ-Al2O3催化剂所含有的Mn4+与Olatt相对较高,可提高反应系统内O3分解能力与污染物反应活性。
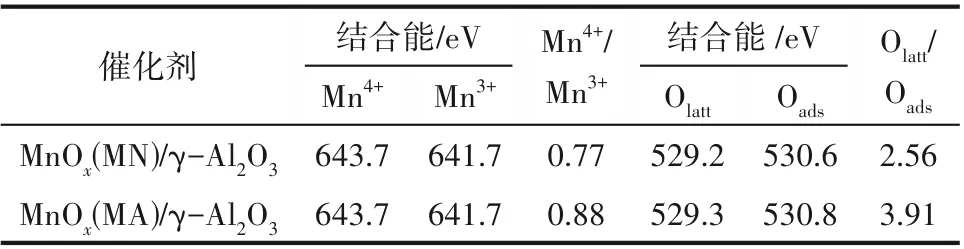
表2 不同前体制备的新鲜Mn基催化剂XPS表征结果Table 2 XPS characterization results of fresh Mn based catalysts prepared from different precursors
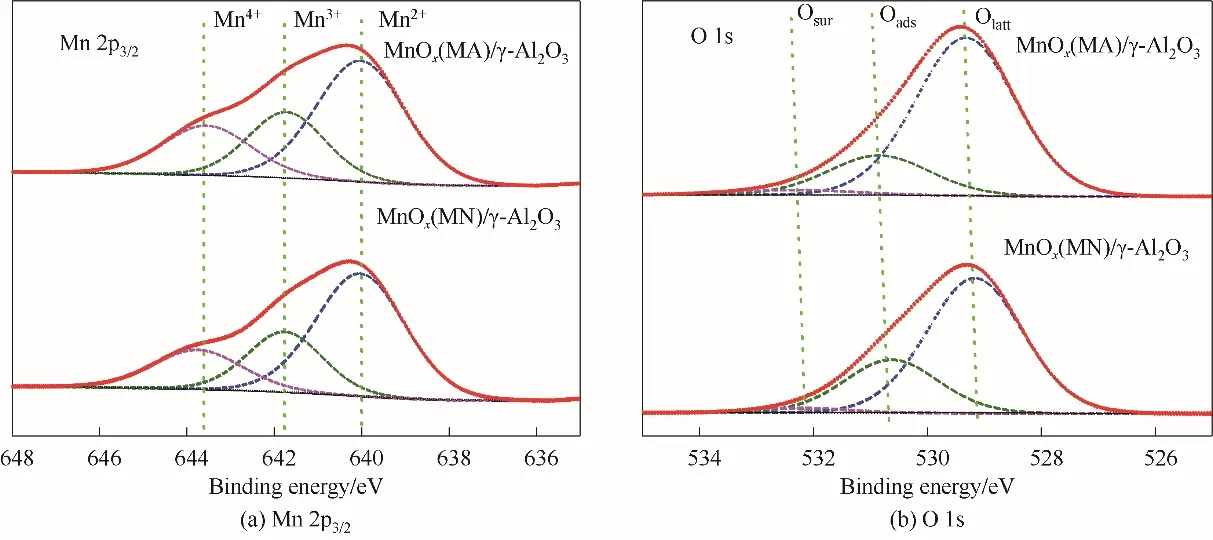
图8 不同前体制备的新鲜Mn基催化剂XPS谱图Fig.8 XPS spectra of fresh Mn based catalysts prepared from different precursors
2.3 产物生成分析
2.3.1 臭氧分析 臭氧是NTP 降解VOCs 过程中不可避免的副产物,同时也是促进VOCs 降解的重要活性物质之一,但其对大气有害,因此需要对臭氧的产生进行控制。
图9为不同电压条件下不同催化剂反应体系中臭氧浓度及反应器表观温度变化情况。如图9(a)所示,对同一反应体系,随着放电电压的增大,臭氧浓度呈现下降趋势,所有反应体系在22 kV 时臭氧浓度基本趋于0;反应器表观温度则呈现上升趋势。相同电压条件下,催化剂的引入降低了反应系统内臭氧浓度,放电电压为16 kV 时,引入γ-Al2O3填料系统的臭氧浓度低于单独NTP反应系统,负载MnOx的填料系统内臭氧浓度低于未负载MnOx的填料系统;在18~22 kV 时,引入填料系统的臭氧浓度基本趋于0。臭氧在常温下分解缓慢,随着温度升高分解逐渐加快,当温度达到100℃时分解剧烈,反应系统内填料的引入增强了放电效应,提高了系统内部温度,在16 kV 时反应器表观温度达100℃以上,导致系统内臭氧分解速率高于生成速率,臭氧浓度整体表现为下降趋势。对于Mn 基催化剂,臭氧主要在催化剂表面发生分解生成活性氧原子[42-43],降低反应系统内臭氧浓度,且臭氧分解产生的活性氧参与到氯苯降解中,从而提高了氯苯降解效率和EE(图3)。
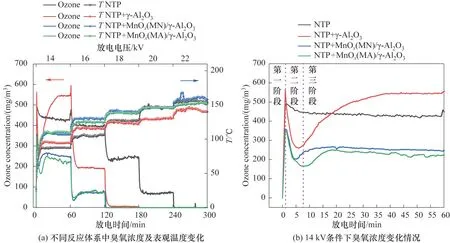
图9 不同电压条件下不同催化剂对臭氧浓度以及反应器表观温度的影响Fig.9 Effects of different catalysts on ozone concentration and reactor apparent temperature under different voltage conditions
在14 kV 时,不同反应系统内臭氧浓度变化规律与其他电压条件下不一致,臭氧浓度变化主要分为三个阶段[图9(b)]:第一阶段为0~1.25 min,臭氧浓度升高,不同反应系统内臭氧浓度增加程度不一致,在放电时间为0.83 min 时,NTP+γ-Al2O3反应系统 内 臭 氧 浓 度 最 高,达 到589 mg∕m3;NTP、NTP+MnOx(MA)∕γ-Al2O3、NTP+MnOx(MN)∕γ-Al2O3反 应 系统均在1.17 min 取得臭氧浓度最大值,分别为491、356 和346 mg∕m3。在NTP 反应系统中,高压放电产生的高能电子与反应系统内O2撞击产生O·,反应系统内填料的填充增强了反应系统内的放电强度(图2),从而导致放电产生的O·浓度增加,反应系统内O·与O2结合生成臭氧,从而导致系统内臭氧浓度呈现升高的趋势,因此,该阶段NTP+γ-Al2O3反应系统臭氧浓度高于NTP 反应系统。而对于NTP+MnOx(MN)∕γ-Al2O3和NTP+MnOx(MA)∕γ-Al2O3反应系统,由于MnOx的存在,部分臭氧在催化剂表面分解为O2和活性氧,臭氧的生成速率大于其分解速率,从而臭氧浓度在整体上呈现上升趋势,但其最高值小于NTP+γ-Al2O3和NTP 反应系统。第二阶段为1.25~7.5 min,该阶段不同反应体系呈现的规律不尽相同。NTP反应系统内,臭氧浓度呈现下降趋势,该现象的出现归结为反应系统内温度升高导致臭氧分解加快,臭氧的分解速率大于其生成速率,从而导致其整体上表现为臭氧浓度下降;NTP+γ-Al2O3、NTP+MnOx(MN)∕γ-Al2O3和NTP+MnOx(MA)∕γ-Al2O3反应系统内臭氧浓度呈现快速下降趋势,随着放电时间的增长,反应系统内温度不断升高,VOCs 分子脱附加快,导致反应系统内VOCs 浓度增加,反应系统内活性氧原子以及生成的臭氧[NTP+MnOx(MN∕MA)∕γ-Al2O3反应系统内部分臭氧在催化剂表面分解为活性氧]与VOCs 分子发生反应,进一步降低了反应系统内臭氧浓度,NTP+MnOx(MN∕MA)∕γ-Al2O3反应系统内臭氧浓度低于NTP+γ-Al2O3反应系统,且NTP+MnOx(MN)∕γ-Al2O3反应系统略高于NTP+MnOx(MA)∕γ-Al2O3反应系统,说明NTP+MnOx(MA)∕γ-Al2O3反应系统内有更多的臭氧在催化剂表面发生分解生成活性氧原子参与到氯苯降解中,从而提高了氯苯降解效率。第三阶段为7.5~60 min,NTP系统呈现基本稳定状态,而填料填充系统内臭氧浓度呈现先上升后平稳趋势,随着放电时间的延长,NTP 系统内VOCs 浓度和温度基本保持不变,该过程中臭氧生成和分解基本维持在相对稳定状态,从而导致系统内臭氧浓度整体呈现平稳趋势[44];而在引入填料系统内,VOCs 继续脱附,但脱附量相对减少,从而导致系统内VOCs 浓度整体呈现减小的趋势,与系统内自由基、臭氧和活性氧的反应速率下降,系统内臭氧浓度上升,当系统内VOCs 浓度和温度达到相对稳定状态时,系统内臭氧呈现相对稳定状态。
基于此,对于不同反应系统,不同放电条件下其臭氧分解原理不一致。填料引入反应系统,在低电压条件下,臭氧浓度不仅受温度影响,而且受填料影响分解为活性氧参与VOCs 的降解;而高电压条件下,臭氧浓度受温度影响更大。
2.3.2 氯元素变化分析 对于低温等离子体降解氯苯,需要对反应过程中的氯元素变化途径进行分析。通过无机氯选择性情况和质谱谱图分析对氯元素变化进行说明,不同反应系统中Cl 选择性(放电电压为14和22 kV)见图10,低温等离子体降解氯苯副产物分析(放电电压为14 kV)见图11。

图10 不同反应系统中Cl选择性(放电电压为14和22 kV)Fig.10 Cl selectivity in different reaction systems(at 14 and 22 kV)
从图10可以发现,在低温等离子体降解氯苯过程中,部分氯元素以无机氯形式存在。分析图11可以发现,反应尾气中物质包括氯苯、含氯有机物、醛类和酮类以及极少数的杂环烷类物质,也可能含有其他种类有机物但未检测到,同时未检测到毒性更强物质,因此,反应过程中氯元素主要分为三类:以氯苯形式存在;以无机氯形式存在;以含氯有机物形式存在。
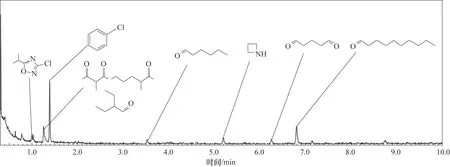
图11 低温等离子体降解氯苯副产物分析(放电电压为14 kV)Fig.11 Analysis of by-products of CB degradation by non-thermal plasma(at 14 kV)
2.3.3 催化剂重复使用稳定性 为探究催化剂抗氯中毒性能,选择相对较优催化剂MnOx(MA)∕γ-Al2O3开展稳定性实验,结果如图12所示。相同条件下开展等离子体降解氯苯实验,催化剂重复使用第3 次时,催化剂降解效率开始出现轻微下降,但未呈现明显的大幅度下降,说明催化剂具有一定的抗性,但要实现实际应用还需进一步研究。
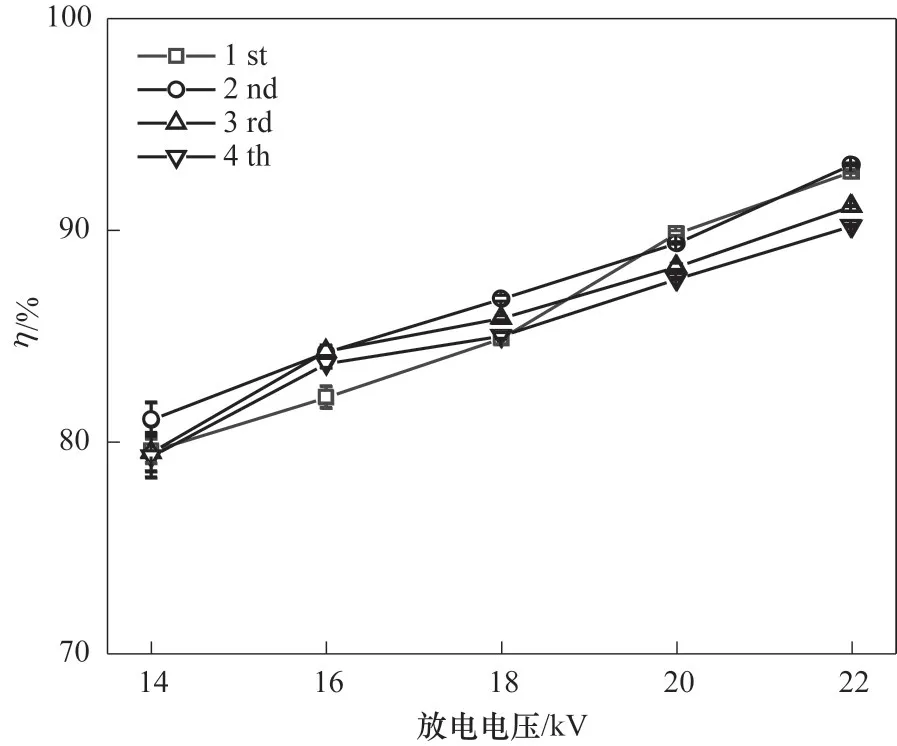
图12 MnOx(MA)∕γ-Al2O3重复用于低温等离子体降解氯苯去除效率变化情况Fig.12 Variation of CB removal efficiency after repeated use of MnOx(MA)∕γ-Al2O3 in NTP
3 结 论
本文研究了NTP 协同催化剂对氯苯降解性能的影响,考察了活性组分Mn 的前体对氯苯降解性能的影响,通过多种手段对反应前后催化剂进行表征分析,同时对不同反应系统的臭氧生成情况进行分析,主要结论如下。
(1)NTP技术能有效降解氯苯,与单独NTP作用相比,填料的引入有效提高了氯苯的降解效率和能量效率,且负载活性组分Mn后其降解性能更佳。
(2)MnOx(MN)∕γ-Al2O3和MnOx(MA)∕γ-Al2O3催化剂均可以提升氯苯的降解效率,且MnOx(MA)∕γ-Al2O3对氯苯降解更好,与MnOx(MN)∕γ-Al2O3相比,MnOx(MA)∕γ-Al2O3具有较大的比表面积,有利于氯苯分子和臭氧的吸附;其表面存在的·OH 和Mnn+为污染物提供了更多的活性位点;活性组分分散更为均匀,有利于催化剂表面臭氧分解为活性更强的活性氧原子,从而提高了氯苯降解性能。
(3)催化剂经放电反应后,其孔径结构和晶相结构不会发生变化。
(4)催化剂引入可有效降低系统内臭氧浓度,负载活性组分Mn 尤其是以乙酸锰为前体后其臭氧浓度下降趋势更为明显,说明在NTP 协同催化剂反应体系中,乙酸锰为前体制备的Mn 基催化剂对臭氧分解能力更好。
(5)低温等离子体降解氯苯过程中氯元素变化形式归为三类,即以氯苯、无机氯和含氯有机物形式存在。
猜你喜欢
产业与科技论坛(2022年18期)2022-10-19
中国资源综合利用(2022年9期)2022-10-13
作物学报(2022年9期)2022-07-18
皮革制作与环保科技(2021年15期)2021-11-10
绿色科技(2021年12期)2021-07-22
绿色科技(2021年4期)2021-04-06
分析仪器(2020年5期)2020-11-09
科学大众(中学)(2019年3期)2019-05-17
中小企业管理与科技·中旬刊(2018年8期)2018-11-12
科技与创新(2015年18期)2015-09-11
