
Non-cell autonomous role of astrocytes in axonal degeneration of cortical projection neurons in hereditary spastic paraplegias
2022-11-11 13:46XueJunLi
中国神经再生研究(英文版) 2022年6期
Xue-Jun Li
Impaired axonal development and degeneration underlie many debilitating diseases, including hereditary spastic paraplegia (HSP). HSPs are a heterogeneous group of neurogenetic disorders characterized by axonopathy of cortical motor neurons (Fink, 2006; Blackstone et al., 2010). The axonal degeneration of these cortical projection neurons (PNs) in HSP patients disrupts the signals from brain to spinal motor neurons, leading to muscle weakness and spasticity. Since the discovery of the first HSP gene (SPAST) in 1999, over 80 distinct genetic loci associated with HSP have been identified. How the mutations of these divergent genes specifically result in axonal degeneration of cortical PNs remains largely unclear, which contributes to the lack of effective treatment to ameliorate, stop, or reverse axonal defects in HSPs.
The most early-onset form of HSP, spastic paraplegia 3A (SPG3A), is caused by mutations in theATL1gene which encodes atlastin-1, a protein critical for the formation of tubule endoplasmic reticulum (ER) threeway junctions. Impaired ER morphogenesis has been observed in SPG3A, as well as other forms of HSP, pointing to a common pathological process in HSP (Rismanchi et al., 2008). However, how ER defects result in axonal deficits remains unclear. Interestingly, it has been recently shown that atlastin-1 and several other common HSP proteins can regulate the size of lipid droplets (LDs) in cultured cellsin vitro(e.g., HEK293, COS7) and in fat bodiesin vivo(e.g., intestinal cells, adipose tissue inC. elegansand flies) (Klemm et al., 2013; Falk et al., 2014; Papadopoulos et al., 2015). These data are in agreement with ER’s role in lipid synthesis and metabolism, implying the involvement of lipid dysfunction in HSP. However, whether lipid metabolism is altered in SPG3A brain and whether lipid dysfunction has any effect on axonal defects of SPG3A cortical PNs are unknown. Using human pluripotent stem cell (hPSC) models of SPG3A, our recent study (Mou et al., 2020) found that lipid droplets are enriched in glial cells, and their sizes are significantly reduced in SPG3A astrocytes. Notably,ATL1mutations result in impaired lipid metabolism and cholesterol transfer in human SPG3A astrocytes, leading to the degeneration of SPG3A cortical PNs. These data demonstrate that astrocytes play a noncell-autonomous role in the degeneration of cortical PNs in HSP.
Studies on SPG3A rely on the use of Drosophila and Zebrafish models which exhibit aberrant nerve outgrowth and locomotion deficits caused by defects in spinal motor neuron axons. There are no SPG3A rodent models that allow examination of cortical PNs directly. We have generated induced pluripotent stem cells from SPG3A patients (Zhu et al., 2014) and differentiated these stem cells into cortical PNs, the cell type affected in patients. We found reduced axonal outgrowth and impaired axonal transport in SPG3A patient stem cell-derived cortical PNs, but not spinal motor neurons, demonstrating the recapitulation of hallmark pathology of SPG3A. Using CRISPR/Cas9-mediated gene editing, we further generated isogenic hPSC lines with two differentATL1mutations (Mou et al., 2020), to examine the mechanisms underlying axonal degeneration of SPG3A cortical PNs. Initial mRNA-sequencing analysis of cortical and spinal cultures revealed specific alterations of genes involved in lipoprotein metabolism and cholesterol trafficking in SPG3A cortical cultures. Remarkably, we found significant lower cholesterol levels in SPG3A cortical PNs compared with control neurons using Filipin staining. The reduced cholesterol levels in SPG3A cultures are further confirmed by examining cholesterol levels using total cholesterol kit. Cholesterol is mainly synthesized in ER and plays important roles in axonal maintenance, repair and neurotransmission (Pfrieger, 2003). Restoration of cholesterol levels in SPG3A cultures can rescue axonal degeneration of SPG3A cortical PNs. Thus, these data reveal that cholesterol deficiency can serve as a novel pathological change implicated in axonal degeneration of SPG3A cortical PNs.Cholesterol is highly enriched in brain, which accounts for 25% of the total cholesterol in the body (Dietschy, 2009). Since the uptake of lipoprotein cholesterol is prevented by blood-brain barrier, cholesterol in the central nervous system (CNS) predominantly comes from de novo synthesis. Glial cells, the major source of cholesterol in the CNS, provide cholesterol to neurons through lipoprotein mediated transfer. Though neurons at early stage during development can synthesize cholesterol, this process gradually reduces after birth and becomes completely dependent on glial cells (Pfrieger, 2003). The specific reduction in the expression of lipoprotein and cholesterol efflux genes in SPG3A cortical cultures suggests the involvement of glial cells in HSP, yet the role of glial cells in HSP is completely unknown. Taking advantage of hPSCs that have the ability to differentiate into various neuronal and glial subtypes, we generated and examined both neurons and astrocytes from control and SPG3A hPSCs. Interestingly, we found that lipid droplets are enriched in astrocytes while much fewer in neurons. The expression of LD genes and the size of lipid droplet were significantly reduced in SPG3A astrocytes, further supporting the implication of astrocytes in the pathogenesis of HSP.
How do mutations of ATL1 in astrocytes result in cholesterol defects in cortical PNs?Our study revealed thatATL1mutations dysregulated proteolipid gene expression including reduced expression of PLIN2, PLIN3, NR1H2 and apolipoprotein E (APOE). APOE is a critical mediator for cholesterol trafficking from glial cells to neurons. The APOE content in the medium was examined using ApoE enzyme-linked immunosorbent assay kit, and a significant reduction of APOE content in SPG3A culture medium was observed. Using cholesterol efflux analysis, we further found that the efflux of cholesterol from SPG3A astrocytes was specifically and significantly reduced compared with control astrocytes (Figure 1).
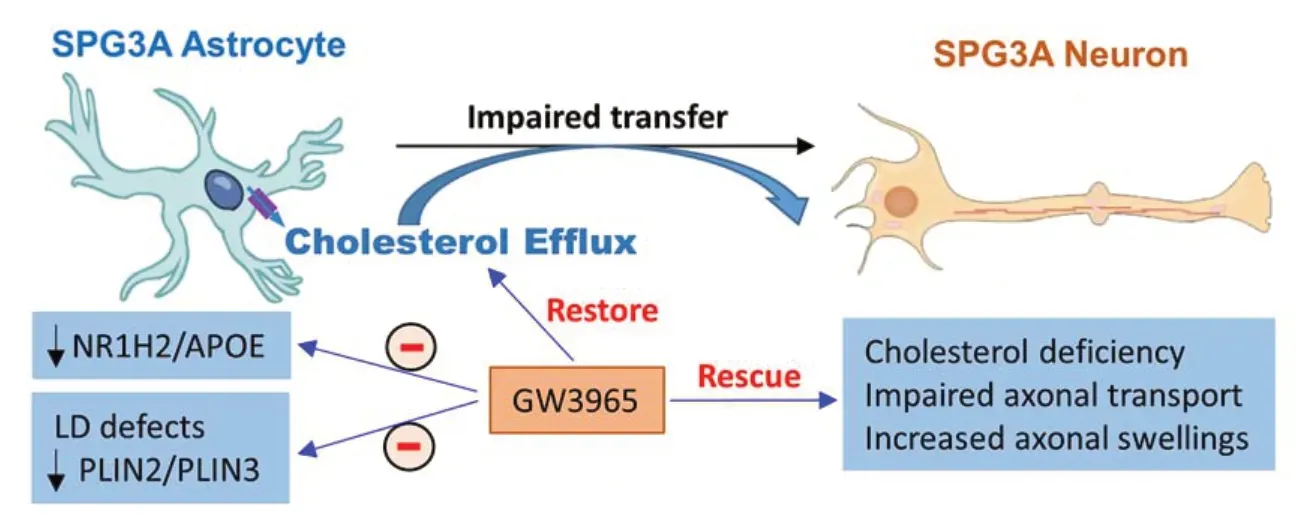
Figure 1|Schematic illustration of impaired lipid homeostasis in SPG3A.
These data reveal thatATL1mutations disrupted cholesterol transfer from glia to neurons, leading to cholesterol deficiency in SPG3A cortical PNs. Considering the reduced expression of NR1H2 (also known as LXRβ), an upstream of ABCA1 and ABCG1 that are mediators for cholesterol efflux, SPG3A cultures were treated with the NR1H2 agonists (e.g., GW3965). The treatment of GW3965 induced the expression of ABCA/ABCG1 and promoted the cholesterol efflux from astrocytes, leading to the restoration of cholesterol levels and the rescue of axonal degeneration in SPG3A cortical PNs (Figure 1). The reduced size of LDs, a direct pathological change caused byATL1mutations in glial cells specifically, was mitigated by the NR1H2 agonist (Figure 1). GW3965 also mitigated the decreased mRNA expression of lipid droplet-related genes (PLIN2andPLIN3), as well asSREBF1, an important regulator for lipid and cholesterol metabolism (Mou et al., 2020). These data together reveal thatATL1mutations impair lipid homeostasis in astrocyte, leading to degeneration of cortical PNs in SPG3A. The role of astrocyte in the axonopathy of HSP is further supported by the rescue of axonal defects in SPG3A cortical PNs after coculturing with control astrocytes, a major source of cholesterol in the brain. Thus, our findings demonstrate a non-cell autonomous mechanism underlying axonal degeneration of SPG3A cortical PNs and identify a new therapeutic target for HSP through regulating cholesterol homeostasis in astrocytes. The detailed mechanisms by whichATL1mutations lead to LD and lipid metabolism defects in astrocytes, as well as alterations of other types of lipids in SPG3A, await further investigation.
Oligodendrocyte is another major lipid producing cell type in the brain, and microglial cells have been reported to regulate lipid trafficking in the CNS. Given that oligodendrocytes and microglial cells are two other key glial cell types along with astrocytes in the CNS, further investigations of two important questions are valuable. First, what are the roles that different types of glial cells play in regulating axonal degeneration of cortical PNs in SPG3A? Protocols to generate various glial cells (i.e., oligodendrocytes, astrocytes, and microglial cells) from hPSCs have been established, providing potential tools to dissect their roles. Second, how do neurons and the interplay between neurons and glial cells contribute to the pathogenesis of HSP? To answer the questions, two challenges need to be addressed. Given that neural cultures derived from hPSCs consist of multiple types of cells, it is critical to enrich or purify different types of neurons and glial cells and then co-culture these cells to dissect their roles and interactions in the pathogenesis of HSP. Another challenge is the difficulty to mimicin vivocircuitry using human stem cell models of disease. Solutions to overcome this issue include building 3-dimension models or transplanting cell into animal models and then examining their maturation and degeneration inin vivoenvironment.
In addition to disease modeling, drug screening is the other important future application of stem cell-derived nerve cells. It has been shown that human cells may have very different responses to drugs compared to other species, and only less than 10% of candidate drugs have been shown to be clinically effective during clinical trials (Kola and Landis, 2004). Human stem cell-derived neurons have been used to test drugs, leading to the identification of more effective compounds (Yang et al., 2013). Therefore, the establishment of systems using hPSCderived neurons to analyze axonal defects will provide a valuable platform to screen therapeutic drugs for HSP. As a proof of concept, in hPSC-based HSP models, we have shown the protective effects of NR1H2 agonist and microtubule-targeting drug against axonal defects (Zhu et al., 2014; Mou et al., 2020). A critical step in developing an effective drug screening system is to identify robust readouts and build reporter lines to measure axonal defects. By combining stem cell technology and gene editing technique, the HSP stem cell disease models and reporter lines will allow a deeper understanding of the pathogenic mechanisms and high-throughput screening of therapeutic agents for rescuing axonal degeneration in these debilitating diseases.
The present work was supported by the National Institutes of Health grant (R01NS118066) and the Blazer Foundation (to XJL).
Xue-Jun Li*
Department of Biomedical Sciences, University of Illinois College of Medicine Rockford, Rockford, IL, USA; Department of Bioengineering, University of Illinois at Chicago, Chicago, IL, USA
*Correspondence to:Xue-Jun Li, PhD, xjli23@uic.edu.
https://orcid.org/0000-0003-1899-9134(Xue-Jun Li)
Date of submission:March 5, 2021
Date of decision:March 29, 2021
Date of acceptance:July 1, 2021
Date of web publication:October 29, 2021
https://doi.org/10.4103/1673-5374.327342
How to cite this article:Li XJ (2022) Non-cell autonomous role of astrocytes in axonal degeneration of cortical projection neurons in hereditary spastic paraplegias. Neural Regen Res 17(6):1265-1266.
Copyright license agreement:The Copyright License Agreement has been signed by the author before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- The importance of fasciculation and elongation protein zeta-1 in neural circuit establishment and neurological disorders
- Promoting axon regeneration in the central nervous system by increasing PI3-kinase signaling
- Microglial voltage-gated proton channel Hv1 in spinal cord injury
- Liposome based drug delivery as a potential treatment option for Alzheimer’s disease
- Retinal regeneration requires dynamic Notch signaling
- All roads lead to Rome — a review of the potential mechanisms by which exerkines exhibit neuroprotective effects in Alzheimer’s disease