
SARS-CoV-2-induced autophagy dysregulation may cause neuronal dysfunction in COVID-19
2022-11-11 13:46MadepalliLakshmana
中国神经再生研究(英文版) 2022年6期
Madepalli K. Lakshmana
The devastating outbreak of the ongoing coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has affected not only the lives of almost everyone around the world but also the governments and societies as well turning into a global catastrophe. Pneumonia of unknown cause was reported in December 2019 in Wuhan, China (Zhu et al., 2020), it rapidly spread to all parts of the globe prompting World Health Organization to declare a pandemic on March 11, 2020 (Cucinotta and Vanelli, 2020). As of October 11th, 2021, 223 countries and territories have reported COVID-19 cases with a total of more than 237 million confirmed cases and more than 4.8 million confirmed deaths (World Health Organization), with the United States leading the world by the highest number of cases thus far (over 43 million infected and 703,599 deaths). Although COVID-19 results mainly in acute lung injury leading to high mortality in the elderly and people with underlying comorbidities, significant nervous system-associated morbidities including meningoencephalitis with elevated lymphocytes and cytokines in the cerebrospinal fluid frequently with viral presence are reported (Lv et al., 2021). Neurons may be more vulnerable to SARS-CoV-2 than SARSCoV because the virus has been confirmed to replicate in neuronal cell lines (Bar-On et al., 2020) and COVID-19 patients show signs of confusion and dizziness which were rarely reported for SARS-CoV. The most troubling outcome of the pandemic is the practice of critical care triage to ration the scarce resources of intensive care units. Therefore, it is very critical to identify how the virus elicits the massive cytokine storm and the failure of homeostatic and defense mechanisms so that mechanism-based novel therapeutics may be identified quickly and lives saved. Here, I propose that cytokine storm which is the major cause of death in COVID-19 patients may indeed be triggered by undigested viral proteins and genomic materials due to viralinduced dysfunction of the autophagy-lysosome pathway (ALP).
Cells have evolved a very complex system of autophagy for eliminating dysfunctional organelles, misfolded proteins, and specifically xenophagy for viral particles and virophagy for viral components which are trapped within an autophagosome and targeted to the lysosome for destruction. Interestingly, autophagic activity is known to decline with age and so far the greatest known risk factor for COVID-19 is age, implying that reduced autophagy may increase the severity in elderly people. Deletion of critical autophagy regulators such as Beclin 1 (BECL1), LC3b, and ATG7 or incubation with a variety of autophagy inhibitors have been shown to markedly enhance secretion of proinflammatory cytokines, suggesting that autophagy plays a pivotal role in regulating inflammation. Thus uncontrolled cytokine storm responsible for disease severity and death in COVID-19 patients may indeed result from SARS-CoV-2-induced autophagy dysfunction. In particular, in the brain, neurons are incapable of self-renewal, are exquisitely susceptible to a wide variety of stress, and therefore for the elimination of viruses, autophagy becomes very crucial. Also, microglia-specific autophagy was shown to be crucial for recovery from neuroinflammation. More importantly, unlike the cerebrum and cerebellum, the detection rate of SARS-CoV-2 was found to be higher in the olfactory system and the brainstem with severe microgliosis and lymphocytic infiltrations (Matschke et al., 2020). Such findings might explain the anosmia and spontaneous loss of breathing frequently seen in severe cases of COVID-19 patients.
As SARS-CoV-2 enters cells through ACE2 receptors facilitated by neuropilin 1 by endocytosis, the host responds first by recognition of specific pathogen-associated molecular patterns (PAMPs) such as spike protein and viral components by pattern recognition receptors, including the RIGI-like receptors, Toll-like receptors and inflammasomes (Li et al., 2020). Pattern recognition receptors can be localized either to cell membranes or intracellularly within various cells of the innate immune system, microglia in the brain, and many epithelial cells to facilitate pathogen recognition. SARS-CoV-2 triggered PAMPS signaling in turn induce a multitude of pathways, including pro-inflammatory responses and autophagy, Interestingly, autophagy also can regulate PAMPS and deliver exogenous viral antigens into MHC-I molecules. Moreover, autophagy can also directly impact the activation, proliferation, and differentiation of virtually all the cell types that participate in adaptive immunity including lymphocytes, antigen-presenting cells, and myeloid cells that contribute to the inflammatory response. Thus autophagy is a critical regulator of both innate and adaptive responses to viral invasion. However, SARS-CoV-2 encodes many virulence factors to evade autophagy or even turn the pathway to their own advantage. The 30 kb genome of SARS-CoV-2 is 100 nm in diameter and contains 14 open reading frames (ORFs) and encodes 24-27 different proteins, including the spike protein, envelope protein, membrane glycoproteins, and nucleocapsid protein (Bar-On et al., 2020). The protein ORF3a of SARSCoV-2 dysregulates the HOPS complex by directly interacting with VPS39 thereby inhibit autophagosome-lysosome (A-L) fusion, a final ALP step critical for efficient degradation of cargo by lysosomes. Interestingly, this property of A-L fusion inhibition through HOPS-VPS39 appears to be specific to SARS-CoV-2 since very similar ORF3a of SARS-CoV was ineffective in inhibiting A-L fusion (Miao et al., 2020). Evidence also suggests that ORF3a through a 20-base sequence also inhibits mitochondrial fusion by targeting USP30 protein, a mitochondrial deubiquitinase that promotes mitochondrial fusion through deubiquitinating ubiquitylated forms of Mfn1 and Mfn2 (Singh et al., 2020). Further, SARS-CoV-2-infected cells express significantly lower levels of MTFP1 and SOCS6 which promote mitochondrial fission, suggesting that ORF3a may also compromise mitochondrial fission ultimately leading to compromise in the mitochondrial quality and early collapse of infected cells. Using affinity-purification coupled to protein identification by mass spectrometry methods, SARS-CoV-2 generated ORF8 protein, as well as nucleocapsid protein, was shown to inhibit mTORC1 by independently binding to mTORC1 pathway molecules La ribonucleoprotein 1 translational regulator and FKBP prolyl isomerase 7 (Gordon et al., 2020). mTORC1, being a negative regulator of autophagy, its SARS-CoV-2-mediated inhibition is expected to activate autophagy. However, although SARSCoV-2-infected cells show autophagosome formation, their maturation appears to be impaired. Remarkably, a Papain-like protease of SARS-CoV-2 has been shown to directly cleave ULK1 at the recognition sequence after G499, separating its N-terminal kinase domain from a C-terminal substrate recognition region and disrupt the formation of ULK1-ATG13 complex (Mohamud et al., 2021). As ULK1 is crucial for autophagosome initiation, its cleavage is expected to stop the entire ALP as there is no autophagosome formation. Thus SARS-CoV-2 has successfully evolved several strategies to circumvent the inherently anti-viral defense capacity of autophagy (Figure 1). Other types of viruses including Coxsackievirus B3 and HIV are known to target TFEB, the master regulator of ALP, and disrupt lysosomal function. Conversely, TFEB overexpression has been shown to increase xenophagy flux and eliminate viruses. It remains to be seen whether SARS-CoV-2 proteins also directly act on TFEB and inhibit ALP. Thus, based on the available evidence SARS-CoV-2 infection in the initial 1-2 weeks may activate autophagy thereby forming more autophagosomes which aid in viral replication, but as more and more viruses are formed and RNAs and proteins get accumulated it may overwhelm the autophagy capacity after 2-5 weeks of infection thereby triggering powerful immune response resulting in cytokine storm and death in vulnerable patients.
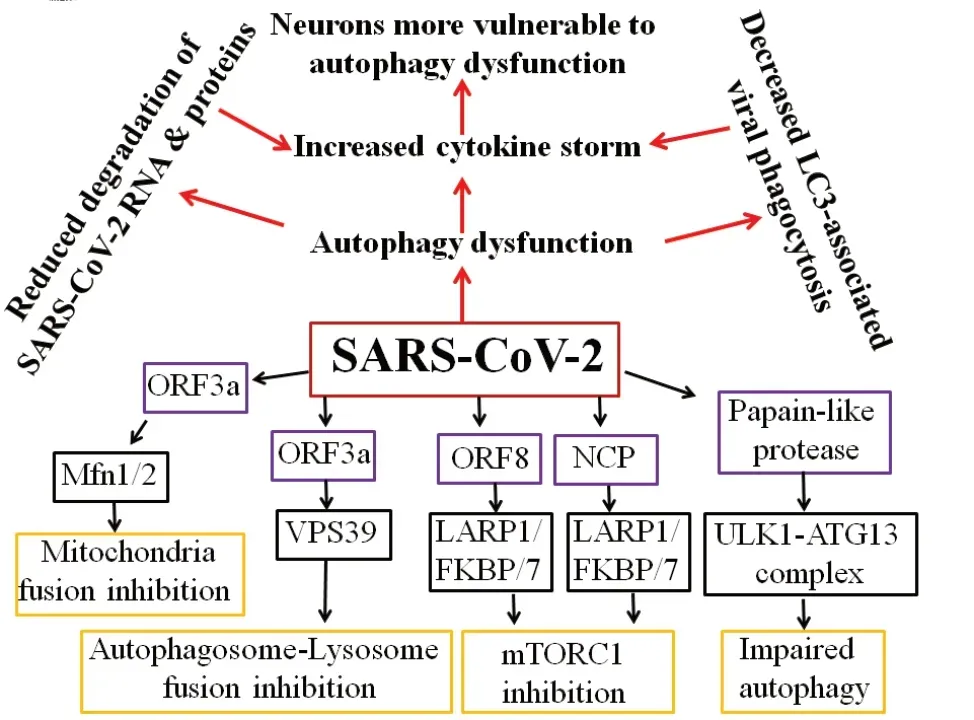
Figure 1|Schematic illustration of the molecular pathways for SARS-CoV-2-induced autophagy dysregulation.
One crucial question that remains unanswered is whether autophagy can be modulated to mitigate SARS-CoV-2-induced massive cytokine storm and its associated toxicity and cell death. Autophagy is the major defense mechanism a cell employs against the pathogens efficiently removes inflammasome-activating stimuli such as PAMPs by recruiting p62 and LC3b and thereby regulates levels of proinflammatory cytokines such as interleukin-6, tumor necrosis factor-α, and interleukin-1β, which are all degraded by autophagy pathway. Another mechanism is through mitophagy, the selective elimination of damaged mitochondria which otherwise would have activated inflammasome and cytokines through released mitochondrial DNA and reactive oxygen species. It is also known that therapeutically increasing autophagy improves survival by reducing the level of inflammation. Thus, autophagy plays a crucial role not only in reducing the viral load but also facilitates tolerance mechanisms that reduce adverse effects of viral infections. Finally, employing the biological activitybased modeling approach, Huang et al. (2021) identified 85 anti-SARS-CoV-2 compounds, among them as many as 35 of them turned out to be autophagy modulators in the GFP-LC3 assay, suggesting that autophagy plays a major role in the antiviral activity of the identified anti-SARS-CoV-2 compounds. There are also several instances where inducing autophagy with a cell-permeable Beclin 1 peptide has been shown to protect against many viruses. Thus, if SARS-CoV-2 induces a massive cytokine storm by suppressing autophagy, its enhancement may have therapeutic implications in COVID-19 patients.
In conclusion, COVID-19 pandemic is a serious ongoing issue worldwide with limited success using vaccinations, but so far no effective antiviral drug has been identified. Although cytokine storm appears to be mainly responsible for causing death, the mechanism by which SARS-CoV-2 elicits massive cytokines is not fully understood. Two independent studies on SARS-CoV-2 protein ORF3a have concluded that although autophagosomes are induced by this protein, their maturation is finally impaired leading to autophagy dysfunction and enhanced inflammation which appears to be a unique feature of SARS-CoV-2. Indeed, a remarkable coincidence between the uncontrolled inflammation triggered by SARS-CoV-2 and autophagy defects have been demonstrated (Garcia-Perez et al., 2020), suggesting that increased cytokine storm may result from the failure of homeostatic regulation by autophagy. However, what is not clear so far is whether autophagy is involved in SARS-CoV-2 protein degradation and whether SARS-CoV-2 evades recognition and degradation by inhibiting A-L fusion at the later period of infection coincident with cytokine storm. Importantly, A-L fusion inhibition appears to be unique to SARS-CoV-2 because a similar ORF3a protein of SARS-CoV failed to inhibit. Specific experiments addressing the temporal sequence of autophagy changes following SARS-CoV-2 infection in specific cell types including neurons and microglia should be designed to answer such questions. This may represent an enormous challenge for the scientific community around the world.
We thank all the administrative and technical staff in the Department of Immunology and Nano-Medicine, Herbert Wertheim College of Medicine (HWCOM), Florida International University, USA, for their administrative and technical support.
This work was supported by a grant from the National Institute of Health (No. 1R21AG060299) to MKL.
Madepalli K. Lakshmana*
Department of Immunology and Nano-Medicine, Alzheimer’s Disease Research Unit, Herbert Wertheim College of Medicine, Florida International University, Miami, FL, USA
*Correspondence to:Madepalli K. Lakshmana, PhD, mlakshma@fiu.edu.
https://orcid.org/0000-0002-1291-4438(Madepalli K. Lakshmana)
Date of submission:March 21, 2021
Date of decision:April 26, 2021
Date of acceptance:July 6, 2021
Date of web publication:October 29, 2021
https://doi.org/10.4103/1673-5374.327333
How to cite this article:Lakshmana MK (2022) SARS-CoV-2-induced autophagy dysregulation may cause neuronal dysfunction in COVID-19. Neural Regen Res 17(6):1255-1256.
Copyright license agreement:The Copyright License Agreement has been signed by the author before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- The importance of fasciculation and elongation protein zeta-1 in neural circuit establishment and neurological disorders
- Promoting axon regeneration in the central nervous system by increasing PI3-kinase signaling
- Microglial voltage-gated proton channel Hv1 in spinal cord injury
- Liposome based drug delivery as a potential treatment option for Alzheimer’s disease
- Retinal regeneration requires dynamic Notch signaling
- All roads lead to Rome — a review of the potential mechanisms by which exerkines exhibit neuroprotective effects in Alzheimer’s disease