
SARS-CoV-2 involvement in central nervous system tissue damage
2022-11-11 13:46MuhammadAliHaidarZaynabShakkourMohammadAmineReslanNadineAlHajPerlaChamounKarlHabashyHasanKaafaraniShimaShahjoueiSarahFarranAbdullahShaitoEsberSabaBassamBadranMirnaSabra0FirasKobeissyMayaBizri
中国神经再生研究(英文版) 2022年6期
Muhammad Ali Haidar, Zaynab Shakkour, Mohammad Amine Reslan, Nadine Al-Haj, Perla Chamoun, Karl Habashy, Hasan Kaafarani, Shima Shahjouei, Sarah H. Farran, Abdullah Shaito, Esber S. Saba, Bassam Badran, Mirna Sabra0,, Firas Kobeissy,,, Maya Bizri,
Abstract As the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) continues to spread globally, it became evident that the SARS-CoV-2 virus infects multiple organs including the brain. Several clinical studies revealed that patients with COVID-19 infection experience an array of neurological signs ranging in severity from headaches to life-threatening strokes. Although the exact mechanism by which the SARS-CoV-2 virus directly impacts the brain is not fully understood, several theories have been suggested including direct and indirect pathways induced by the virus. One possible theory is the invasion of SARS-CoV-2 to the brain occurs either through the bloodstream or via the nerve endings which is considered to be the direct route. Such findings are based on studies reporting the presence of viral material in the cerebrospinal fluid and brain cells. Nevertheless, the indirect mechanisms, including blood-clotting abnormalities and prolonged activation of the immune system, can result in further tissue and organ damages seen during the course of the disease. This overview attempts to give a thorough insight into SARS-CoV-2 coronavirus neurological infection and highlights the possible mechanisms leading to the neurological manifestations observed in infected patients.
Key Words: autoantibodies; CNS infection; coagulopathy; COVID-19; encephalitis; neuroinflammation; renin-angiotensin system; viral encephalopathy
Introduction
Today, hundreds of viral pathogens, including rhinoviruses, influenza viruses, parainfluenza viruses, adenoviruses, and coronaviruses, amongst others, have been implicated in respiratory illnesses (Vareille et al., 2011; Desforges et al., 2019). Many of these viruses circulate year-round, while others exhibit seasonal patterns (Desforges et al., 2019; Moriyama et al., 2020). Occasionally, certain viral strains, initially confined to animal reservoirs, successfully acquire human tropism and these usually constitute the most imminent threats to human health (Taubenberger and Kash, 2010). Coronaviruses (CoVs), from the Coronaviridae family (Weiss and Navas-Martin, 2005), best exemplify this phenomenon. Indeed, this family of viruses was found to infect humans since the early 1960s and was since described as harmless with self-limiting symptoms, until 2003 when severe acute respiratory syndrome coronavirus 1 (SARS-*CoV-1) emerged. SARS-CoV-1 is the causative agent of severe acute respiratory syndrome (SARS) (Ye et al., 2020) and its origin was linked to palm civets (Li et al., 2006).
On the 29thof December, 2019, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), originally named (2019-novel CoV or 2019-nCoV), was identified as the causative agent of coronavirus disease 2019 (COVID-19). SARSCoV-2 is a member of the Betacoronavirus genus (Mao and Jin, 2020; Ye et al., 2020). In fact, its closest related viruses, by sequence identity of the RNA genome, are bat-SL-CoVZC45 (87.99% sequence identity, 99% query coverage) and bat-SLCoVZXC21 (87.23% sequence identity, 98% query coverage), both of which are from bat origin. Its nomenclature, however, refers to the more distant SARS-CoV-1, with which it shares 79% sequence identity (Zhou et al., 2020).
As of February 2021, more than 100 million COVID-19 cases have been confirmed worldwide, with over 2 million cases resulting in fatalities. Commonly experienced symptoms include fever, dry cough, myalgia, anosmia, and respiratory distress (Organization; Mao and Jin, 2020). More serious presentations involve acute respiratory distress syndrome (ARDS) (Haidar et al., 2021), systemic inflammatory response syndrome (Masi et al., 2020), and multisystem inflammatory syndrome (Jiang et al., 2020). Asymptomatic cases are also prevalent, but not without drawbacks. Asymptomatic carriers remarkably contribute to the rapid spread of the disease and hinder national and international containment efforts (Gao et al., 2020b).
For the most part, the human-human spread of SARS-CoV-2 is through respiratory droplets, but transmission through fomites or aerosols has also been reported, under certain circumstances (Jayaweera et al., 2020; Wang and Du, 2020). The virus initially infects the host’s respiratory tract, where it is exposed to alveolar epithelial cells, macrophages, and vascular endothelial cells (Harrison et al., 2020). Viral attachment and entry into host cells are primarily mediated by the viral “Spike” (S) protein, made up of two fused protein subunits termed S1 and S2 (Harrison et al., 2020). During viral infection of the host cell, the receptor-binding domain (RBD) of the S1 subunit first binds to the host angiotensin-converting enzyme 2 (ACE-2) receptor, mediating cellular attachment. Cell entry is then activated by the proteolytic cleavage of the S protein subunits at the S1/S2 cleavage site by host cell proteins cathepsin L and transmembrane protease serine 2 (TMPRSS2), exposing a fusion peptide in the S2 subunit (Harrison et al., 2020). The tissue tropism of SARS-CoV-2 and its mechanism of infection are similar to that of SARS-CoV-1. However, crucial mutations in SARS-CoV-2 RBD have favored more potent interactions with the host’s ACE-2 receptors, accounting for its superior infectivity (Harrison et al., 2020; Yan et al., 2020).
Along the same line, respiratory viruses, including CoVs, have long been able to infect the brain tissue (Bergmann et al., 2006; Desforges et al., 2019). Evidence of the effects of SARSCoV-2 on the central nervous system (CNS) is evolving, with the virus being linked to neurological as well as psychiatric symptoms (Varatharaj et al., 2020). Several studies found COVID-19 to be associated with neurological manifestations in as high as 36% of the patients (Mao et al., 2020). When neurological and neuropsychiatric symptoms were reported, the most common manifestations were cerebrovascular events (62%), followed by altered mental status (31%) (Varatharaj et al., 2020). Neurological manifestations can range from a mild headache (Mao et al., 2020) or “brain fog” (Nauen et al., 2021), to more serious complications such as Guillain-Barre syndrome (Alberti et al., 2020), encephalitis (Moriguchi et al., 2020), and arterial and venous strokes (Kananeh et al., 2020). Some of the earliest reports of CNS involvement also described an unusually high rate of anosmia and dysgeusia (Varatharaj et al., 2020).
Viral invasion of the CNS tissue has been postulated following the identification of SARS-CoV-2 RNA in the cerebrospinal fluid of symptomatic patients (Moriguchi et al., 2020). The postulated mechanisms of invasion are numerous and have been extensively reviewed by Ladecola et al.(2020). Similar to other tissues, brain blood vessels express ACE-2 receptors (Hamming et al., 2004), which serve as docking sites for the virus, leading to its hematogenous dissemination. The frequently experienced anosmia has also raised the possibility of a direct invasion through the olfactory bulb (Iadecola et al., 2020). Indeed, ACE-2 receptors and TMPRSS2 have been identified in the nasal mucosa (Brann et al., 2020; Nampoothiri et al., 2020), and MRI hyperintense signals suggestive of infection have been detected in the olfactory bulbs of patients with COVID-19 (Politi et al., 2020). The virus could therefore attach to olfactory nerve terminals, get internalized by endocytosis, and then get transported to other brain regions, as will be further discussed in relation to the direct effects of the virus on CNS tissues (Iadecola et al., 2020). This is directly relevant to the ongoing debate over the cause of COVID-19 damage to nervous tissues. While there is acceptable evidence concerning the neurological manifestations related to SARS-CoV-2 infection, data about the presence of the virus in cerebrospinal fluid are conflicting (Al Saiegh et al., 2020; Destras et al., 2020). These complications are becoming concerning especially with the development of different vaccines which have raised questions regarding their contribution to certain thrombotic events within the CNS. Hence, this review will attempt to expose the possible mechanisms of how the virus can affect brain tissue either through its direct presence in the brain tissue or indirectly. Also, these mechanisms will be associated with how the brain damage leads to several complications that await further investigation.
Search Strategy
For this review, we searched the published literature (EMBASE, MEDLINE, PubMed, CINAHL) and grey literature from their inception dates to 2021. For possible ongoing trials and studies, we searched ClinicalTrials.gov registry and examined reference lists of published narratives and systematic reviews and abstracts from relevant scientific meetings. We used broad text and MeSH terms to maximize sensitivity.
Database search was conducted using a mixture of keywordbased search and controlled vocabulary search strategies. Key concepts were identified as MeSH and combined with key terms as follows:
1. “Renin-Angiotensin System/physiology”[Mesh]) AND (“COVID-19”[Mesh] OR “SARS-CoV-2”[Mesh]);
2. (COVID*[tw] OR SARS-CoV-2[tw] OR “SARS-CoV-2”[Mesh]) AND ((“Central Nervous System”[Mesh]) OR (brain[tw]) OR (central[tw] nervous[tw] system*[tw]) OR (spinal[tw] cord[tw]));
3. (COVID*[tw] OR SARS-CoV-2[tw] OR “SARS-CoV-2”[Mesh]) AND (“Neurogenic Inflammation”[Mesh] OR neuroinflamm*[tw]).
Mechanisms of SARS-CoV-2 Pathogenesis
Direct mechanisms: ACE-2 and Kallikrein kinin system
Most of the data about SARS-CoV-2 interaction with host cells are based on similarities drawn from the interaction of the S protein of SARS-CoV-1 with the host cell ACE-2 receptor (Li, 2015). A recent analysis of sequence identities between SARS-CoV-1 and SARS-CoV-2 suggested that the two strains are akin in their binding to ACE-2 (Wan et al., 2020; Zhang et al., 2020a), with the latter being more efficient in recognizing human ACE-2 (Nguyen et al., 2020; Wrapp et al., 2020). This may help to understand the rapid spread and easier transmissibility of SARS-CoV-2 and its current pandemic state. In this regard, Zhou et al. (2020) have shown that SARS-CoV-2 can enter ACE-2-expressing cells but not cells that lack ACE-2 expression, confirming that ACE-2 is the host cell receptor for SARS-CoV-2. Using single-molecule force spectroscopy, Yang et al. (2020) demonstrated that the S1 subunit and the RBD of SARS-CoV-2 S protein can bind with high intrinsic affinity to the ACE-2 receptor.
ACE-2 is a crucial element of the renin-angiotensinaldosterone system (RAAS), which is involved in blood pressure regulation, electrolyte homeostasis (Bourgonje et al., 2020), cellular growth and proliferation, inflammation, as well as extracellular matrix remodeling (Nehme et al., 2019). ACE-2 genetic variants have been under investigation in relation to the increased risk of SARS-CoV-2 infections, but have not yet been assessed in large samples in order to determine if certain genetic variants are risk factors for developing certain SARS-CoV-2 symptoms (Shovlin and Vizcaychipi, 2020). While ACE is found ubiquitously expressed in the vasculature, ACE-2 expression, in comparison is restricted to endocrine tissues, lung alveolar epithelial cells (type I and type II), small intestinal epithelial cells, cardiovascular tissue, testis, muscle cells, and the brush border of proximal tubular cells (Bourgonje et al., 2020; Wang et al., 2020). In particular, ACE-2 was shown to protect mice from acute lung injury induced by acid aspiration or sepsis (Imai et al., 2005), suggesting that ACE-2 is not only the viral entry point for coronaviruses but also participates in an important lung-protective pathway. This protective pathway is usually dysregulated in the event of SARS-CoV-2 infection (Zhang et al., 2020a).
The RBD region is a class I fusion protein responsible for the fusion of host and viral membranes in a process driven by S protein conformational changes (Zhang et al., 2020a). Upon viral binding, Gln 394 in the RBD region of SARS-CoV-2 interacts with Lys 31 of the human ACE-2 receptor (Yan et al., 2020). Following the binding of the RBD to ACE-2, TMPRSS2 cleaves the S1/S2 cleavage site, to expose a fusion peptide in S2, facilitating membrane fusion. Then, the viral ssRNA(+) is released into the cytoplasm of the host cell, and a set of sub-genomic RNA is transcribed. Six open reading frames that can serve as a template for the synthesis of sub-genomic mRNA have been identified in the SARS-CoV-2 genome. While it is established that ACE-2 is the receptor used by the virus to infect human cells, other factors are often used by CoVs to maximize their infectious potential (Hoffmann et al., 2020). In addition to TMPRSS2 and cathepsin L, neuropilin-1 is suggested to serve as another host factor for SARS-CoV-2 infection. Indeed, using x-ray crystallography and biochemical analysis, it was shown that the R motif at the C-terminal of S1 binds to neuropilin-1 at the cell surface (Daly et al., 2020). In confirmation, RNA interference or selective inhibition of this interaction demonstrated a reduction in SARS-CoV-2 entry and infectivity in cell culture (Daly et al., 2020). Additional host cell entry factors have been suggested, such as CD209 and CD209L, but are yet to be confirmed (Amraie et al., 2020).
Other than facilitating viral entry, the major contribution of ACE-2 to the pathogenesis of COVID-19 is through its participation in the RAAS system. The RAAS system involves enzymatic cascades that instigate the conversion of angiotensinogen secreted by the liver into the active angiotensin II (Ang-II) by renin, an aspartyl protease that cleaves angiotensinogen into the vaso-inactive angiotensin I decapeptide, Ang-I (1-10) (Ribeiro-Oliveira et al., 2008; Sparks et al., 2014). This product is then processed, by the exopeptidase of membrane-bound ACE, to produce the vasoactive octapeptide Ang-II (1-8). Ang-II signaling through its receptor AT1R is involved in vasoconstriction in addition to other detrimental roles such as inflammation, fibrosis, and alteration of cellular redox states. Ang-II AT2R signaling, on the other hand, is vasodilatory and has regenerative and protective functions being antifibrotic and anti-inflammatory (Nehme et al., 2019). The hyperactivation of RAAS has been reported to induce inflammation due to the release of inflammatory and profibrotic cytokines such as transforming growth factor-beta, increased vascular permeability, and hyperactivation of the complement cascade, including C5a and C5b-9.
Another major product of the RAAS system is the Ang (1-7) peptide. Ang (1-7) is generated by ACE-2 acting either directly, Ang-II (1-8) converted into Ang (1-7), or indirectly, Ang (1-7) produced from Ang (1-9). In the latter case, ACE-2 converts Ang-I (1-10) into Ang (1-9) which is then converted by ACE into Ang (1-7) (Donoghue et al., 2000; Nehme et al., 2019). Ang (1-7) is next secreted into the circulation and tissues such as those of the heart, kidney, liver, and blood vessels where it exerts local paracrine and autocrine actions (Donoghue et al., 2000; Nehme et al., 2019). Ang (1-7) acts mainly through a G-protein-coupled receptor, Mas (Simões e Silva and Teixeira, 2016). Mas receptor activation by Ang (1-7) can cause apoptosis, reduction in cell proliferation (Costa-Neto et al., 2014), and fibrosis (Chappell and Al Zayadneh, 2017), and can have protective effects against lung injury (Li et al., 2016). Ang (1-7) also promoted brain angiogenesis by attenuating the reduction of cerebral blood flow following ischemia, leading to an improved stroke outcome (Jiang et al., 2014). Notably, Ang (1-7) is reported to have a protective effect against hyperactivation of the RAAS, by virtue of its antagonistic effects on the ACEAng-II/AT1R axis. For instance, overactivation of the ACE/Ang-II/AT1R axis participates in the maintenance and development of hypertension, an important risk factor for ischemic stroke, possibly through inhibition of oxidative stress and inflammation (Jiang et al., 2013).
Along the same line, a large body of evidence suggests that Ang (1-7) is the biologically active component of RAAS, which gained recognition after the discovery that ACE-2 was the major enzyme contributing to its formation (Santos et al., 2018) where it converts Ang-II to Ang (1-7), which in turn binds to its receptor Mas (Jiang et al., 2013). In light of that, Ang (1-7), has been shown to have a modulatory role in the brain, especially in the brainstem and hypothalamus, where it exerts a synergistic and antagonistic effect on Ang-II (Soltani Zangbar et al., 2021). ACE-2 acts to limit brain renin-angiotensin system hyperactivity, through interaction between Ang AT1a receptors and ACE-2 in the brainstem (Ribeiro-Oliveira et al., 2008). The ACE-2/MAS axis normally inhibits the proinflammatory actions of ACE/Ang-II/AT1R axis by reducing the levels of Ang-II- through the processing of Ang-II to produce Ang-(1-7) and converting Ang I, the Ang-II precursor, into Ang (1-9)- and reducing the expression of nuclear factor kappa B (NF-κB), mitogen-activated protein kinases (MAPKs), and inflammatory factors interleukin-8 (IL-8), tumor necrosis factor α (TNF-α), and IL-6 (Mahmudpour et al., 2020). Relatedly, upon infection, SARS-CoV-2 is seen to downregulate the expression of ACE-2 on cells, resulting in elevated levels of soluble ACE-2 in the blood, urine, and other body fluids. This may lead to an imbalance between the renin-angiotensin system and ACE-2/angiotensin-(1-7)/MAS receptor axis, possibly contributing to multiple organ injuries in COVID-19 (Furuhashi et al., 2020). This is mainly achieved by activation of the NF-κB inflammatory pathway, leading to an increase in the production of proinflammatory mediators TNF-α, IL-1β, IL-6, and IL-10, which are usually observed in moderate to severe cases of COVID-19 (Soltani Zangbar et al., 2021).
ACEs also have a fundamental role in the Kallikrein kinin system (KKS) where kininogens are cleaved into high and low molecular weight kininogen by Hageman factor and/or trypsin in the plasma and tissues, ultimately giving rise to two kinins: bradykinin (BK) in plasma (product of the breakdown of high molecular weight kininogen) and kallidin in tissues (lysyl-BK; a product of the breakdown of the low molecular weight kininogen) (Carvalho et al., 2021). These two kinin peptides can activate bradykinin B2 receptors (BKB2R), resulting in increased vascular permeability (Mugisho et al., 2019), nociceptor activation (Vellani et al., 2004), and vasodilation (Sigurdsson et al., 2014). In addition, BKB2R signaling can activate proinflammatory pathways through stimulation of nitric oxide and prostaglandin synthesis (Sigurdsson et al., 2014; Carvalho et al., 2021). SARS-CoV-2 can express a cysteine protease that activates the kinin pathway by interacting with kininogens (Solowiej et al., 2008), NF-κB, complement cascades, mediators of apoptosis, and a number of pattern recognition receptors such as toll-like receptors (Mahmudpour et al., 2020). Kinins can activate another receptor, BKB1R, which is strongly upregulated by inflammatory mediators such as cytokines, leading to a positive feedback loop between BKB1R and cytokines as well as an increase in the endothelial permeability and leukocyte migration (Zheng et al., 2020). As a result kinins/BKB1R signaling may generate what is known as a “cytokine storm” characterized by sepsis-like symptoms (Carvalho et al., 2021). Importantly, BK has a half-life of barely seconds in the plasma, where ACE has been demonstrated to cleave BK by removing its carboxyl-terminal to generate BK(1-7), an inactive peptide (Cyr et al., 2001). In context, TMPRSS2 also cleaves kininogen and activates the production of bradykinin (Lalmanach et al., 2010).
Markedly, a local elevation of cytokines in the cerebral tissue can induce inflammation, leading to neurological features similar to those encountered in SARS-CoV-2 patients (Soltani Zangbar et al., 2021). One suggested mechanism for the neurological symptoms following SARS-CoV-2 infection is the disruption of gas exchange by the respiratory system, leading to general hypoxia and causing an anaerobic metabolism in the mitochondria that leads to an overproduction of acid. This leads to ischemia, cerebral swelling and blood flow obstruction, vasodilation, and headaches, especially in highrisk patients (Wu et al., 2020). In line with this, postmortem examination of the brains of COVID-19 patients showed brain tissue edema, partial neuronal degeneration, and vascular and infection-related secondary inflammatory tissue damage due to abnormal immune response (Fisicaro et al., 2021). However, it is still unclear whether these findings are a direct viral pathology or a non-specific consequence of COVID-19 on the brain.
Nevertheless, it is still unclear whether the virus can enter the brain despite the direct presence of the virus in the CNS has been documented in some studies. In fact, evidence suggests that SARS-CoV-2 may have neuro-invasive potential, which could be associated with significant brain damage, neurological diseases, and damage to the medullary cardiorespiratory center, which may, in turn, affect respiratory function (DosSantos et al., 2020). Effectively, the olfactory pathway offers the strongest candidate route for SARS-CoV-2 invasion of the CNS. In fact, there is a crosstalk between the olfactory pathway and subcortical and cortical structures that may explain the olfactory disturbances that frequently arise in SARS-CoV-2 patients. Of particular importance is the fact that olfactory receptor neurons are replaced throughout life, unlike other mammalian nerves. These neurons are continuously regenerated from undifferentiated basal cells, which explains why SARS-CoV-2-related olfactory symptoms may not be permanent (DosSantos et al., 2020). The olfactory pathway starts at the upper part of the nasal cavity/olfactory mucosa, where the olfactory epithelium is located. The olfactory receptors are located on bipolar neurons that form small bundles that comprise the olfactory nerve (DosSantos et al., 2020). Such anatomical features allow CoVs to reach the brain and cerebrospinal fluid through the olfactory nerve and bulb within 7 days, causing inflammation and demyelinating reaction (Bohmwald et al., 2018). Interestingly, ACE-2 was found to be expressed in mouse olfactory bulb, human piriform cortex, and some hypothalamic nuclei in the human brain (Chen et al., 2021). The exact mechanism through which SARS-CoV-2 reaches the CNS via the olfactory pathway is still unclear, yet when the virus enters brain tissue, it initiates a cascade of inflammatory events that have been linked to brain tissue swelling, cerebral blood flow obstruction, interstitial edema, and cerebral vasodilation (Wu et al., 2020). These findings align with the notion that inflammation is one important etiology to the CNS complications induced by the virus such as headaches due to congestion and ischemia, ischemic stroke, projectile vomiting, and visual loss (Wu et al., 2020), as will be further discussed.
Indirect mechanisms: neuroinflammation
Neuroinflammation is a complex innate immune response induced by neural tissue to repair cellular damage, limit infection, or eliminate pathogens. It can be either positive or negative and may be followed by a variety of physiological, biochemical, and behavioral consequences (DiSabato et al., 2016). Typically, acute neuroinflammation, characterized by an early and short inflammatory response, is considered neuroprotective and is initiated by the activation of glial and endothelial cells (Shastri et al., 2013). Prolonged activation, on the other hand, is detrimental to the CNS. In fact, chronic neuroinflammation has been associated with several neurodegenerative diseases including Alzheimer’s disease (AD), Parkinson’s disease (PD), and multiple sclerosis (Kempuraj et al., 2016). This aforementioned inflammatory response is mediated by glial cells in the CNS. Specifically, microglia are the resident innate immune cells responsible for the regulation of neuroinflammation and exhibit a multitude of functions in the CNS including stimulus-specific microglial response (Shastri et al., 2013; Wu et al., 2015; Colonna and Butovsky, 2017). In the event of pathogenic infection, microglia become activated through the binding of the exogenous pathogen-associated molecular pattern to pattern-recognition receptors on their surface. Once activated, they shift their transcriptional profile to produce proinflammatory cytokines including TNF-α, IL-1β, and IL-6; as well as chemokines and reactive oxygen species in order to counteract the infection (Konat et al., 2006; Kraft and Harry, 2011). Similarly, the astrocytes, considered the most abundant glial cells in the CNS, play a crucial role in regulating innate and adaptive immune response, and contribute to the maintenance and permeability of the blood-brain barrier (BBB). Activated astrocytes undergo various molecular and morphological changes depending on the context of stimuli (Sofroniew, 2009). Inflammatory cytokines such as IL-1β, TNF-α, and interferon (IFN)-γ are major inducers of astrocytic activation (Xu et al., 2020).
Interestingly, it has been shown that the activation of microglia and astrocytes is a key mediator of the neuroinflammatory response induced via SARS-CoV-2 infection (Tremblay et al., 2020). For instance, Kanberg et al. (2020) investigated glial activation in patients with moderate and severe COVID-19 by measuring plasma biomarkers of CNS injury. Neurofilament light chain protein was also found to be significantly increased in patients with severe infection (P< 0.001), whereas glial fibrillary acidic protein was elevated both in patients with severe (P= 0.001) and those with moderate infection (P= 0.03). Another case report documented an increase in glial fibrillary acidic protein expression in the white matter of a patient who died from COVID-19 (Reichard et al., 2020). Microglial cells are considered a major source of cytokine production including interleukins (ILs) and INFs involved in the “cytokine storm” in the CNS (Lavi and Cong, 2020). Cytokine storm is defined as an unregulated immune response due to an auto-amplified cytokine production (Tang et al., 2020). Such event is in part induced by IL-6 amplifier, which once stimulated, would hyper-activate the machinery responsible for regulating the NF-κB pathway (Hojyo et al., 2020). Since cytokines can contribute directly to neuronal damage (Zimmermann et al., 2017), it has been suggested that microglial activation can be a mediator of the neurological manifestations of COVID-19 infection (Vargas et al., 2020). Among the deleterious effects of the cytokine storm is the initiation of cytokine release syndrome which has been associated with high mortality in COVID-19 patients (Hirano and Murakami, 2020; McGonagle et al., 2020). For instance, based on the cytokine profiles from COVID-19 patients, several studies reported that the cytokine storm was directly correlated with multi-organ failure, lung injury, and other unfavorable prognoses of severe infection (Chen et al., 2020; Gao et al., 2020a; Ruan et al., 2020; Sun et al., 2020). The brain, however, is more susceptible to the cytokine storm partly due to “microglial priming” (Mishra and Banerjea, 2020) Neuroinflammation is a significant contributor to this phenomenon that involves the change of microglial proliferation, morphology, physiology, and phenotype. Microglial priming induces an exaggerated microglial response in CNS that is more sensitive to minor stimuli (Li et al., 2018), including peripheral cytokines. Consequently, a positive feedback loop is attained in which “primed microglia” produces more cytokines and inflammatory mediators that further disrupt the CNS homeostasis with a major impact on synaptic plasticity and neuronal survival (Li et al., 2018; Bouayed and Bohn, 2021). Several studies performed on patients that passed away from COVID-19 reported the presence of microgliosis and microglial nodules (Schurink et al., 2020; Bertram et al., 2021; Lee et al., 2021). In another post-mortem study by Matschke et al. (2020), a high degree of astrogliosis was observed as well. The authors of the study suggested that microgliosis and astrogliosis in the olfactory bulb might be responsible for the anosmia commonly observed in COVID-19 patients.
SARS-CoV-2 and Brain Damage
The above mechanisms exhibit a complex interplay as they loop into one another leading to the neurological manifestations seen in COVID-19. This interplay makes the distinction between the direct and indirect effects of SARSCoV-2 unclear. Assuming successful entry of the virus into brain tissue, its interaction with the ACE-2 receptor would activate a cascade of events leading to the cytokine storm. In this regard, overactivation of RAAS, acting through ACE/Ang-II/AT1R arm, can induce inflammation through profibrotic cytokines like TGF-β (Mahmudpour et al., 2020). As it has been eluted to earlier, SARS-CoV-2 can lead to downregulation of ACE-2 causing a reduction of Ang (1-7) production and as a result removal of the brake on the ACE/Ang-II/AT1R signaling leading to uncontrolled hyperactivation of the RAAS system. As a result, the NF-κB pathway is activated leading to elevated production of proinflammatory cytokines IL-6, TNFα, IL-1β, and IL-10 (Pacurari et al., 2014; Mahmudpour et al., 2020). The release of these molecules and their accumulation in the serum leads to cytokine storm and eventually to acute respiratory distress syndrome (ARDS) in severe cases (Figure 1) (Mahmudpour et al., 2020).
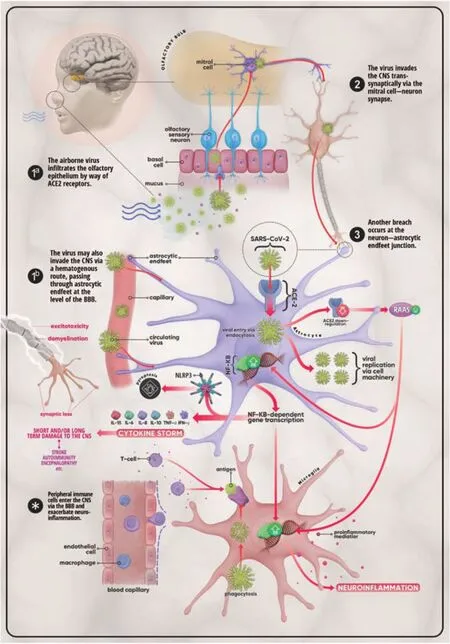
Figure 1|Mechanisms of invasion and possible CNS effects of SARSCoV-2.
Additionally, the purinergic ionotropic P2X7 receptor signaling has been implicated in the buildup of the cytokine storm. P2X7 receptor signaling effectively stimulates NLRP3 inflammasomes, leading to IL-1β and IL-18 release, and caspase-1 activation (Di Virgilio et al., 2020). This pathway can offer an innovative understanding of the inflammatory state during COVID-19 infections and consequently is the focus of an intensive investigation. During the early tissue damage caused by the disease, adenosine triphosphate (ATP), an important damage-associated molecular pattern, is released and activates its surface receptors known as P2 receptors. P2X7 receptor belongs to the P2X ligand-gated ion channel receptors subfamily of P2 receptors. The P2X7/NLRP3 pathway stimulates K+efflux and pyroptosis, an inflammation-mediated cell death that employs several caspases and the cleavage of the protein gasdermin D (Soltani Zangbar et al., 2021). The details of this pathway were studiedin vitroin epithelial cells where elevated ATP levels increased cytotoxicity and production of inflammatory cytokines, nitric oxide, and reactive oxygen species in mouse macrophages (Mishra, 2013). P2X7/NLRP3 not only helps in creating an inflammatory microenvironment but also plays a role in amplifying chemotaxis signaling and controlling cell orientation. This is also the case in neutrophil activation upon damage where ATP acts in an autocrine manner to modulate the activation of P2X7 and vascular cell adhesion molecule-1 (VCAM-1) release, a neutrophil chemoattractant that promotes tissue infiltration (Soltani Zangbar et al., 2021). These cumulative effects, driven by the different pathogenic pathways related to COVID-19, have given rise to a myriad of neurological complications that complicate the COVID-19 disease.
Encephalopathy
The risk of developing encephalopathies in COVID-19 patients is relatively high (Filatov et al., 2020) and can stem from several COVID-19 complications. To start with, an elevation of blood pressure (BP) can cause cerebral hyper-fusion and eventually brain edema leading to the BBB breakdown. In this case, autoregulatory mechanisms that protect the BBB from dangerous systolic BP increases in a normal state becomes disrupted (Immink Rogier et al., 2004). Besides, acute elevations of BP cause orthostatic plasma leakage over the capillaries which, combined with BBB breakdown, initiates a cascade of events that may lead to hypertensive encephalopathy (Schwartz et al., 1992). In COVID-19 patients, this notion should be seriously considered since hypertension is often reported during SARS-CoV-2 infections and the virus reduces ACE-2 expression, increasing the risk of Ang-II dependent hypertension (Wysocki et al., 2010; Lippi et al., 2020).
Also, hypoxemia has become a hallmark feature of COVID-19. As a result, encephalopathy can also proceed through another route initiated by the hypoxic state manifested during the infection (Garg et al., 2021). Hypoxic-ischemic brain injury (HIE) or encephalopathy, can result from hypoperfusion of oxygen and low supplies of glucose in the brain causing anaerobic metabolism in cerebral tissues (Chauhan et al., 2020; Wu et al., 2020). The resultant low cellular pH evokes intracerebral vasodilation, brain edema, obstruction of cerebral blood flow, and ischemia. The basal ganglia, hippocampal formation, cerebellum, and thalamus, are the brain regions between the anterior and middle cerebral arteries that have been reported to be specifically and greatly affected by HIE (Peng Zhou, 2020). In the case of COVID-19, one study encompassing 257 patients, 9% of the patients presented with altered mental status suggestive of encephalopathy (Cummings et al., 2020). Another study by Scullen et al. (2020) showed retrospective data of 27 critically ill patients, 74% of who were diagnosed with COVID-19-associated encephalopathy based on neuroimaging. One review analyzed 42 records from 56 isolated cases and suggested that encephalopathy/encephalitis was more common in older patients, noting that these were severely ill (Garg et al., 2021). Further investigation of the mechanisms leading to encephalopathies is crucial considering the widespread report of encephalopathies among COVID-19 patients.
Stroke
Although pulmonary effects are the main concern in COVID-19 patients and ARDS is the leading cause of death by this virus, cardiovascular symptoms and complications are not to be overlooked. Cardiovascular problems such as stroke, thromboembolism, deep vein thrombosis, and myocardial infarctions have increasingly become an important cause of death in SARS-CoV-2 infection (Haidar et al., 2021). The risk of developing a stroke differs between races (Ikewaki et al., 2020) and increases with age, comorbidities, and severity of infection (Iadecola et al., 2020). However, multiple stroke cases were presented in young COVID-19 patients, including previously healthy ones, which raises the hypothesis that the virus itself may be responsible for the initiation and exacerbation of thrombotic events (Oxley et al., 2020). Changes in the vasculature that accompany COVID-19 infection can predispose patients to a stroke. This discussion will focus more on ischemic strokes as they appear more frequently during COVID-19, but it is worthy to mention that the virus may potentially increase the risk for hemorrhagic strokes as well. Hemorrhagic strokes can be a result of the damage evoked by the virus in the intracranial vessels, which express the ACE-2 receptor, and their subsequent rupture; this is in addition to blood vessel injury initiated by systemic inflammation and the cytokine storm (Spence et al., 2020).
The factors that may contribute to thrombosis during COVID-19 are numerous. Since ACE-2 receptors are widely expressed by endothelial cells, blood vessels become an important target of the virus, leading to endothelial injury and eventually to thrombotic events. The depletion of ACE-2 receptors by SARS-CoV-2 counteracts the normally protective function of this receptor and leads to endothelial injury (Qi et al., 2020). A post-mortem study conducted on COVID-19 patients found viral inclusion structures, lymphocytic endotheliitis, endothelial cell apoptosis, and accumulation of inflammatory cells in the vascular endothelium (Varga et al., 2020). This demonstrates the direct injury of endothelial cells by the virus and the inflammation that accompanies the infection.
Two major causes of the blood stasis observed in COVID-19 patients are hyper-viscosity and hyper-coagulability. The former is a result of the cytokine storm and the increase in fibrinogen levels (Qi et al., 2020). Hyper-viscosity, assessed by viscometry, was seen in 15 critically ill COVID19 patients (range, 1.9 to 4.2 centipoise (cP); normal range, 1.4 to 1.8 cP) (Maier et al., 2020). Furthermore, immobilization, which is known to increase the risk of thrombus formation, is another factor that contributes to blood stasis in hospitalized COVID-19 patients (Maier et al., 2020). To add, another study found that some severely ill patients have increased levels of anti-phospholipid antibodies which are associated with thrombosis (Qi et al., 2020).
On the other hand, COVID-19-associated hyper-coagulability is due to the systemic inflammation and cytokine storm characterized by the high levels of IL-6 and the tissue factors that initiate the coagulation cascade (Qi et al., 2020). Notably, SARS-CoV-2 infection is accompanied by coagulation abnormalities such as increased D-dimer formation, increased levels of fibrinogen and von Willebran factor (VWF), and factor VIII activity (Panigada et al., 2020). In context, a study conducted on 24 selected patients with severe COVID-19 pneumonia showed that these patients have increased fibrin generation, greater clot strength, reduced fibrinolysis, and a small decrease in antithrombin, demonstrating the contribution of hyper-coagulability to COVID-19 (Panigada et al., 2020). Additionally, Zaid et al. (2021) demonstrated that a resistant hypercoagulable state in COVID-19 patients is mediated by platelet hyperreactivity. The study found that this state was noticeably increased in patients with COVID-19 compared with patients who have ARDS unrelated to COVID-19.
Some features of COVID-19 coagulopathy are similar to those found in disseminated intravascular coagulation. However, some reports contradicted these findings. For example, elevated D-dimer levels with a lack of thrombocytopenia and a relatively normal prothrombin time, partial thromboplastin time, and antithrombin levels show that the coagulopathy of COVID19 is different from disseminated intravascular coagulation (Iba et al., 2020). Besides, the high fibrinogen and factor VIII are also contradictory with a typical coagulopathy where coagulation factors are consumed (Panigada et al., 2020). Importantly, the consistent patterns observed in some coagulation parameters, like an increase in D-dimer and fibrinogen levels, show promise as a prognostic parameter for infection outcomes. For example, D-dimer levels were uniformly associated with the severity of the infection, suggesting that it may be a serve as a prognostic marker, especially when combined with IL-6 (Tomo et al., 2021). However, some studies demonstrated contradictory results in the case of fibrinogen. One study found that fibrinogen levels were higher in COVID-19 patients; however, others noticed that fibrinogen levels did not correlate with the severity of COVID-19 disease (Zhou et al, 2020; Tomo et al., 2021). It is worthy to mention here that thrombotic events have also been described post-vaccination against SARS-CoV-2, albeit not being related to the virus itself. Attention towards this subject became of interest in late 2020 when two patients developed myelitis after taking the Oxford/AstraZeneca vaccine (Goss et al., 2021). Starting from March 2021, the Vaccine Adverse Event Reporting System (VAERS) presented data showing that around 17 cases of stroke have been reported postvaccination (Goss et al., 2021). Another case report described two young males who developed severe thrombocytopenia and fatal cerebral venous sinus thrombocytopenia after being administered a COVID-19 vaccine (Mehta et al., 2021). Although no causational relationship has been described between vaccines and thrombotic events, it is important nonetheless to further investigate their effects and pinpoint any underlying mechanisms that could be at play.
Epilepsy
New-onset seizures have recently been reported in COVID-19 patients, although it is not a common presentation. Seizures took place as focal motor, tonic-clonic, convulsive status epilepticus, or non-convulsive status epilepticus (Asadi-Pooya et al., 2021). Many medical centers failed to extensively test COVID-19 patients who developed seizures, further limiting available data on SARS-CoV-2 and epilepsy (Asadi-Pooya et al., 2021). Interestingly, one case report describes a focal status epilepticus as the initial presentation in a COVID-19 patient with the absence of pneumonia and prodromal symptoms (Vollono et al., 2020). The 78-year-old woman was already predisposed to epilepsy due to postencephalitic epilepsy. However, her MRI, 10 days before admission was normal and her medications controlled her state and kept her seizurefree for 2 years until she was infected with SARS-CoV-2 (Vollono et al., 2020). Rare cases of de novo seizures have also been documented. A healthy 73-year-old COVID-19 patient presented with recurrent focal seizures and was diagnosed with focal epilepsy (Elgamasy et al., 2020). Another case study reported that a COVID-19 patient with new-onset multiple generalized tonic-clonic seizures (Karimi et al., 2020).
Furthermore, in a Chinese cohort study conducted on 304 COVID-19 patients, none of the individuals developed nonconvulsive status epilepticus (Lu et al., 2020). However, seizure-like events were observed in two cases. One patient had bilateral body spasms with mouth deviation and no alternation of consciousness. She was diagnosed with an acute anxiety disorder which was properly treated. The other patient had bilateral myoclonus in the limbs, also with no impairment in consciousness. However, these spasms were caused by hyponatremia, hypokalemia, and hypocalcemia and were resolved when the electrolyte values were back to normal (Lu et al., 2020).
The mechanism by which these seizures developed is unclear, but it is hypothesized that they are either due to direct injury of CNS by the virus or secondary due to the increase in inflammatory cytokines, which then can activate glutamate receptors causing hyperexcitability of the nervous system and acute seizures (Karimi et al., 2020). In this regard, elevated IL-6 levels during COVID-19 can possibly contribute to seizure occurrence since elevated IL-6 levels were previously associated with febrile seizures (Li et al., 2011). along the same line, intranasal administration of IL-6 in rat models showed exacerbation of seizures (Vohora et al., 2020). Additionally, COVID-19 associated complications like hypoxia, metabolic disturbances, strokes, and cytokine storms may also participate in seizure development (Lu et al., 2020).
Autoimmunity and Neurodegeneration in SARS-CoV-2
Another COVID-19 complication that can have consequences in the CNS is the production of autoantibodies. This complication raises major concerns when studying the long-term effects associated with COVID-19. Indeed, autoantibodies, unlike cytokine storms, are believed to cause targeted and long-term damage in COVID-19 patients (Khamsi, 2021). Recent studies have shown that antibodies to SARSCoV-2 can cross-react with host proteins of blood vessels, heart, and brain, contributing to certain clinical phenotypes seen in infected patients (Mohammadi et al., 2020; Pfeuffer et al., 2020; Franke et al., 2021; Guilmot et al., 2021). Reported cross-reactivities with host proteins in severe SARSCoV-2 infections include autoimmunity against INFs type I, phospholipids, and annexin A2 (Bastard et al., 2020; Woodruff et al., 2020; Zuo et al., 2020; Wang et al., 2021; Zuniga et al., 2021). Bastard et al. (2020) showed that approximately 101 of 987 patients with severe COVID-19 pneumonia had autoantibodies against INF type I molecules, leading to a block of its action which usually boosts the immune response against foreign pathogens. In contrast, out of 663 individuals with an asymptomatic or mild infection, none had these autoantibodies. This implies that the detection of autoantibodies could be one approach that can predict the severity of COVID-19. Nevertheless, the authors reported that 4 of 1227 healthy individuals had such autoantibodies before the viral infection, suggesting that genetic predisposition might play a role in the production of these autoantibodies (Bastard et al., 2020). Interestingly, Zhang et al. (2020b), using the candidate gene approach, examined the genetics underlying life-threatening COVID-19 and identified an association between severe infections and mutations in genes involved in the regulation of type I and type III IFN immunity. The authors concluded that individuals at high risk of lifethreatening infection can be identified and that in the early stages, administration of type I INFs may be therapeutically useful in some patients. The correlation between autoantibodies against IFN and increased risk of infectious disease was first suggested by Pozzetto et al. in 1984. Likewise, autoantibodies against phospholipids and phospholipidbinding proteins, which are involved in regulating blood clotting, can result in unfavorable side effects. A recent study showed that 52% of 172 patients hospitalized with COVID-19 had anti-phospholipid antibodies and that such autoimmunity may put the patients at higher risk of thrombotic arterial and venous occlusions (Zuo et al., 2020). Similarly, autoimmunity against annexin A2, a phospholipid-binding protein that helps in the maintenance of the stability of cell membranes and the integrity of pulmonary microvasculature, was studied among patients with COVID-19. Zuniga et al. (2021) found that the levels of anti-annexin A2 antibodies were significantly higher in people who died of COVID-19 than in those with non-critical illness (P= 0.006). Since the inhibition of annexin A2 induces systemic thrombosis and cell death, the authors indicated that elevated levels of such autoantibodies could predict mortality. A recent cohort study of 194 COVID-19 patients used Rapid Extracellular Antigen Profiling to screen for autoantibodies against 2770 extracellular and secreted proteins (Wang et al., 2021). The researchers found that in comparison to uninfected controls, patients display a noticeable increase in autoantibody reactivity with the predominance of autoantibodies against immunomodulatory proteins such as complement components, cytokines, chemokines, and cell surface proteins. Analysis of such autoimmunity has shown a correlation with certain clinical characteristics and disease severity. The autoantibody theory raises the question as to whether the onset of severe symptoms in COVID-19 is evoked by the virus itself or is a secondary outcome of the autoimmune response induced by the infection.
Several studies suggested that autoantibodies found in patients with COVID-19 can reach the brain (Delamarre et al., 2020; Franke et al., 2021; Guilmot et al., 2021; Mulder et al., 2021). Autoimmune neurological syndromes associated with the COVID-19 include Guillaine-Barré syndrome (GBS): a postinfectious autoimmune disorder characterized by progressive weakness in the lower extremities. The onset of GBS varies in duration from one day before the appearance of COVID-19 symptoms to 3 weeks later (Paterson et al., 2020; Toscano et al., 2020). This autoimmune response is mediated by molecular mimicry in which antibodies can cross-react with host gangliosides (Ebrahim Soltani et al., 2019). Interestingly, besides ACE-2 receptors, the binding of COVID-19 spike protein to host cells can occur through sialic acid-containing glycoproteins and gangliosides. Typically, antibodies to the ganglioside GD1b (anti-GD1b) result in ataxic neuropathy (Dalakas and Quarles, 1996). A recent study has shown that such antibodies found in the serum of COVID-19 triggered GBS patients implicate the cross-reactivity of COVID-19-binding gangliosides and peripheral nerve glycolipids (Dalakas, 2020). Furthermore, Zhao et al. (2020) suggested that the COVID-19-mediated cytokine storm and the consequent BBB disruption and lymphocyte infiltration can contribute to the development of GBS. Other post-infectious autoimmune neurological disorders, including meningitis and encephalitis, have also been reported (Brun et al., 2020; Dixon et al., 2020; Grimaldi et al., 2020; Moriguchi et al., 2020).
On an additional note, patients recovered from COVID-19 infection may develop adverse cognitive complications indicating possible brain injury and a higher risk of dementia including AD. There as several molecular events occurring during COVID-19 infection that are significantly implicated in AD, primarily neuroinflammation (Daugherty et al., 2020; Naughton et al., 2020; Zhou et al., 2021). Recently, Zhou et al. (2021) constructed a network-based, multimodal COVID-19 virus-host interactome using protein-protein interaction assay and CRISPR-Cas9 genetic results to show evidence between the viral infection and dementia-like symptoms. Based on the results from PPI and CRISPR-Cas9 genetic assays, the authors found that COVID-19 host proteins and genes significantly affected the expression of proteins and genes associated with AD including RAB7A, TGFB1, and VCAM1. In addition, aberrant expression of AD biomarkers, such as NKTR, GSTM3, TGFB1, TNFRSF1B, SPP1, and CXCL10, were detected in the cerebrospinal fluid and blood of patients with COVID-19 . The authors of the study concluded that there is a significant mechanistic overlap between COVID-19 infection and AD (Zhou et al., 2021).
Likewise, recent studies suggest a correlation between COVID-19 infection and PD (Brundin et al., 2020; Cohen et al., 2020; Faber et al., 2020; Méndez-Guerrero et al., 2020; Merello et al., 2021). So far, three cases of Parkinsonism after COVID-19 infection have been reported (Cohen et al., 2020; Faber et al., 2020; Méndez-Guerrero et al., 2020). The three patients (aged 35, 45, and 58 years) developed clinical PD within 2-5 weeks post COVID-19 infection. Interestingly, brain imaging showed classic signs of PD although none of them had a sign of prodromal PD or a family history of PD. One patient performed genetic testing revealing the absence of any PD risk variants (Cohen et al., 2020). In two of three patients, symptoms of PD were reduced when given traditional dopaminergic medication (Cohen et al., 2020; Faber et al., 2020) and the third recovered without medication (Méndez-Guerrero et al., 2020). Brundin et al. (2020) discussed possible mechanisms that could explain the appearance of Parkinsonism post COVID-19 infection. For instance, vascular complications and inflammation induced by COVID-19 infection are hallmarks related to the progression of PD. Furthermore, the detection of viral RNA in postmortem brains of some patients with COVID-19 suggests that the neuroinvasive nature of the virus could be a potential factor associating the viral infection with the development of PD (Brundin et al., 2020).
The short and long-term effects of COVID-19 on the brain are not yet fully known; however, enough evidence proves its potential as a risk factor for brain injury and neurodegeneration. Prospective longitudinal studies are crucial for better understanding the lasting implications of COVID-19 on the progression of neurological disorders including AD and PD.
Ultimately, looking into the long-term effects of COVID-19 is important to understand the pathways not only related to the pathology, but also regeneration. The first step in doing so is investigating the persistent symptoms that take place after the acute infection takes place. For example, in a meta-analysis that looked at neuropsychiatric symptoms 6 months after the infection from 51 studies, the authors found that cognitive impairments, known as brain fog, were reported at 20.2% (Badenoch et al., 2021). Sleep problems were at 27.4% and post-traumatic stress disorder was at 15.7%. For this reason, post-mortem andin vivostudies are especially essential to look at temporal changes in inflammation and other pathways related to CNS damage to assess their relationship with recovery and long-term pathology. This is especially important because inflammation poses one important element that affects regenerative outcomes after CNS-related traumas (Benowitz and Popovich, 2011). Previous studies enticing peripheral nerve graft and injection of a proinflammatory glucan molecule derived from the yeast cell wall showed retinal ganglion cell axonal growth in rodents (Leon et al., 2000; Lorber et al., 2005). Interestingly, some molecules that act on Toll-like receptor 2 such as Pam3Cys have been shown to stimulate axonal regeneration, pointing at the potential for inflammatory pathways to be involved in neuroregeneration (Hauk et al., 2010).
Conclusion
The pathoetiology behind SARS-CoV-2 related neurological complications has been the focus of many recent studies. The pathogenesis of this infection starts with its direct entry into CNS tissue through the olfactory bulb by binding to the ACE-2 receptor, and inducing inflammatory cascades via the RAAS and KKS systems. Its interaction with the immune system and activation of innate immunity cascades is another important pathway involved in the development of the cytokine storm, a major concern in COVID-19 mortality. The complex interaction between these two mechanisms characterizes SARS-CoV-2 involvement in neurological complications which include encephalitis, stroke, epilepsy, and autoimmunity. Further understanding of these different manifestations may provide preventative measures to curb COVID-19 transmission and offer novel therapeutic avenues in the treatment of COVID-19.
Acknowledgments:Special thanks go to Ms. Leila Nasrallah, MS; and Ms. Yara Yehya, MS, American University of Beirut, for their generous efforts in reviewing and editing the manuscript.
Author contributions:Conceptualization: FK, MAH, and ZS; supervision: FK, MB, MS, and BB; original drafting: MAH, ZS, MAR, NAH, PC, KH, and HK; manuscript review and editing: SS, AS, ES; illustrations: MAR.
Conflicts of interest:The authors declare no conflicts of interest.
Financial support:This work has been partially funded by a joint grant funded by the Flash Grant Call from the National Council for Scientific Research in Lebanon (CNRS-L) and the American University of Beirut (AUB) titled: “Neurological Complications PostCoronavirus Disease (COVID-19)“ (to FK) and “SARS-COV2 antibodies signature in exposed individuals to assess protection status” (to ES).
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- The importance of fasciculation and elongation protein zeta-1 in neural circuit establishment and neurological disorders
- Promoting axon regeneration in the central nervous system by increasing PI3-kinase signaling
- Microglial voltage-gated proton channel Hv1 in spinal cord injury
- Liposome based drug delivery as a potential treatment option for Alzheimer’s disease
- Retinal regeneration requires dynamic Notch signaling
- All roads lead to Rome — a review of the potential mechanisms by which exerkines exhibit neuroprotective effects in Alzheimer’s disease