
抗GD2单克隆抗体药物的质量控制
2022-11-08 03:16杜加亮于传飞王文波崔永霏郭璐韵杨雅岚俞小娟
山西医科大学学报 2022年9期
杜加亮,于传飞,王文波,武 刚,崔永霏,郭璐韵,杨雅岚,俞小娟,李 萌,王 兰
(中国食品药品检定研究院单克隆抗体产品室,卫生部生物技术产品检定及标准化重点实验室,国家药品监督管理局生物制品质量研究与评价重点实验室,北京 102629;#共同第一作者;*通讯作者,E-mail:wanglan@nifdc.org.cn)
单克隆抗体在肿瘤患者临床治疗中的作用受到越来越广泛的关注和重视,我国也将部分单克隆抗体纳入医保[1]。2021年美国食品药品管理局(Food and Drug Administration,FDA)批准了第100个单抗制品[2],预计2022年单抗的全球销售额将高达2 000亿美元[3]。随着新靶点、研发管线和生物类似药的不断增加,以及对单抗作用机制研究的不断深入[4],监管部门对于单抗制品的质量控制也提出了越来越高的要求。
神经母细胞瘤(neuroblastoma,NB)是儿童最常见的颅外实体肿瘤,其中高危NB患儿中仍有一半以上在经过各种治疗后终因复发而死亡[5]。自20世纪80年代以来,科学家陆续发现双唾液酸神经节苷脂(disialoganglioside,GD2)在几乎所有的NB中高表达,而在正常人组织中的表达有限,因此GD2作为癌症免疫治疗的主要靶标具有很高的价值,也被美国国家癌症研究所列为75种潜在抗癌靶标中的第12位[6]。经过40多年的发展,目前已有3种抗GD2单抗获得FDA或欧洲药品管理局(EMA)的批准上市[7-9],用于高危NB患儿的临床治疗。我国仅有一种抗GD2单抗于2021年获批上市,另一种正在审评中。
伴随着该单抗制品在我国高危NB患儿临床治疗中的使用,将有越来越多的针对该靶点的原研药和生物类似药的研发。因此,为了有效地对抗GD2单抗进行质量控制,保证其安全性和有效性,本研究依据现行版《中华人民共和国药典》和人用药品注册技术要求国际协调会议(The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use,ICH)的指导要求[10,11],研究建立抗GD2单抗的生物学活性、结合活性、纯度分析、电荷异质性分析、糖基化分析等方法,针对产品的关键质量属性进行分析和探讨,为抗GD2单抗的质量控制提供依据和指导。
1 材料与方法
1.1 抗体和主要试剂
重组人源化抗GD2单克隆抗体(以下简称抗GD2单抗)及其参比品为中国食品药品检定研究院单克隆抗体产品室留存。人神经母细胞瘤细胞LA-N-1和补体购自德国Merck公司。重组人GD2抗原购自美国Santa Cruz公司。重组人CD16a抗原购自加拿大R&D SYSTEMS公司。二硫苏糖醇(DTT)、8.0 mol/L盐酸胍、色谱级甲酸和乙腈购自美国ThermoFisher Scientific公司。碘乙酰胺(IAM)购自美国AMRESCO公司。载体两性电解质Pharmalyte 3-10、8-10.5及氨偶联试剂盒购自美国GE Healthcare公司。pI标志物5.85及9.22、1%甲基纤维素溶液、0.5%甲基纤维素溶液、cIEF柱FC-涂层均购自美国ProteinSimple公司。三氟乙酸(TFA)、乙二胺四乙酸四钠盐水合物(EDTA)、2-甲基吡啶硼烷、2-氨基苯甲酰胺、甲酸铵、遗传霉素、2-巯基乙醇、N-乙基马来酰亚胺(NEM)、一水合磷酸二氢钠、无水磷酸氢二钠、无水硫酸钠及5%叠氮化钠溶液均购自美国Sigma公司。谷氨酰胺、胎牛血清、细胞培养基RPMI-1640购自美国Gibco公司。N-糖苷酶F、质谱级胰蛋白酶、Bio-Glo荧光素酶检测试剂盒、Cell Titre-Glo试剂盒、ADCP报告基因检测试剂盒购自美国Promega公司。
1.2 LA-N-1细胞杀伤试验测定抗GD2单抗的补体依赖性细胞毒性(complement-dependent cytotoxicity,CDC)作用
取对数生长期的LA-N-1细胞用测定培养基悬浮,并调至细胞密度约为0.75×106个/ml,以25 μl/孔加入96孔板;用测定培养基稀释样品,以90 μg/ml为起始浓度,按照1 ∶4,1 ∶4,1 ∶3,1 ∶3,1 ∶3,1 ∶3,1 ∶4,1 ∶4的比例(V/V)依次稀释(共9个稀释度,包括起始浓度),以25 μl/孔加入对应孔中,37 ℃,5% CO2培养15~30 min;取补体工作液(10 ml测定培养基稀释2.3 ml补体)以25 μl/孔加入相应孔中,吹打混匀至少5次,37 ℃,5% CO2培养4~5 h;取平衡至室温的CellTitre-Glo溶液,以75 μl/孔加入对应孔中,100~150 r/min室温避光震荡2~5 min裂解细胞,室温避光孵育10~30 min,使用荧光酶标仪读板。采用四参数法,以样品浓度的对数为横坐标,以平均吸收值为纵坐标,计算半效浓度(EC50,ng/ml)。
1.3 荧光素酶报告基因法测定抗GD2单抗的抗体依赖性细胞介导的细胞吞噬(antibody-dependent cell-mediated phagocytosis,ADCP)效应
取对数生长期的LA-N-1细胞用测定培养基悬浮,并调至细胞密度约为0.4×106个/ml,以25 μl/孔加入96孔板;用测定培养基稀释样品,以3 μg/ml为起始浓度,按照1 ∶2,1 ∶2,1 ∶2,1 ∶1.5,1 ∶1.5,1 ∶2.3,1 ∶2,1 ∶2的比例依次稀释(共9个稀释度,包括起始浓度),以25 μl/孔加入对应孔中;取液氮保存的ADCP效应细胞于37 ℃水浴1 min,用测定培养基将细胞密度调为0.4×106个/ml,以25 μl/孔加入对应孔中,吹打混匀至少4次,37 ℃,5% CO2培养过夜;取平衡至室温的Bio-Glo溶液,以75 μl/孔加入对应孔中,室温避光孵育10~30 min,使用荧光酶标仪读板。采用四参数法,以样品浓度的对数为横坐标,以平均吸收值为纵坐标,计算半效浓度(EC50,ng/ml)。
1.4 酶联免疫吸附试验(enzyme-liked immunosorbent assay,ELISA)测定抗GD2单抗的GD2结合活性
取2 μg/ml GD2工作液,以50 μl/孔加入高结合的96孔板中,于30 ℃真空离心机中完全干燥。后加入封闭液300 μl/孔,于2~8 ℃封闭过夜。以500 ng/ml为起始浓度,按照1 ∶2倍比稀释样品,共11个稀释度,以100 μl/孔至对应孔中,37 ℃孵育60 min。取200 ng/ml的检测抗体工作液,以100 μl/孔加入,室温孵育60 min。取TMB底物溶液以100 μl/孔加入,室温孵育10 min。以100 μl/孔加入终止液,用酶标仪以650 nm波长为参比波长,在波长450 nm处测定吸光度。
1.5 采用SPR-Biacore法测定抗GD2单抗的CD16结合活性
样品自320.89 nmol/L稀释浓度起,以2.8倍系列稀释至0.24 nmol/L(共8个稀释度)。取50 μg/ml抗His抗体工作液偶联420 s(30 μl/min)。CM5芯片使用EDC/NHS溶液(1 ∶1)活化420 s(10 μl/min),使用1 mmol/L乙醇胺(pH 8)去活化420 s(10 μl/min)。分析参数:使用系列S传感器芯片CM5、1×HBS-EP+运行缓冲液、再生溶液(甘氨酸pH 1.5 10 mmol/L)、稀释液1×HBS-EP+、FcγRIIIA(V)浓度1 μg/ml、配体捕获接触时间和速率分别为24 s和10 μl/min,分析物接触时间和速率分别为120 s和30 μl/min、再生接触时间和速率分别为60 s和30 μl/min。
1.6 还原/非还原十二烷基硫酸钠毛细管凝胶电泳(capillary electrophoresis-sodium dodecyl sulfonate,CE-SDS)法进行纯度分析
用超纯水将样品稀释至10 mg/ml,取该溶液10 μl,加入SDS样品缓冲液75 μl,另外,还原CE-SDS法加入巯基乙醇5 μl,非还原CD-SDS法加入0.1 mol/L N-乙基马来酰亚胺5 μl,混匀,之后在72 ℃水浴条件下孵育8 min,室温放置5 min,涡旋混匀并离心之后取90 μl置于进样瓶中后上机分析。使用Beckman PA800 plus毛细管电泳系统,无涂层毛细管(内径50 μm,总长度31 cm,有效长度21 cm)检测。检测条件:进样电压为5.0 kV,分离电压为15 kV,毛细管温度为25 ℃,样品室温度为15 ℃,紫外检测波长为220 nm。计算样品轻链峰+重链峰(还原型)或主峰(非还原型)峰面积百分比以及片段峰面积百分比。
1.7 分子排阻色谱(size exclusion-high performance liquid chromatography,SE-HPLC)法分析样品纯度
取浓度为2~5 mg/ml的样品20 μl,采用Waters e2695高效液相色谱系统配备紫外检测器、Acquity UPLC BEH200 SEC色谱柱(1.7 μm,4.6 nm×300 mm)进行检测。流动相由0.1 mol/L磷酸盐缓冲液和0.25 mol/L Na2SO4配制而成,pH为6.8,流速为0.4 ml/min,上样量20 μl,进样器温度5 ℃,柱温30 ℃,在波长280 nm处检测,使用面积归一化法计算单体和聚合物的百分比。
1.8 电荷异质性分析
使用毛细管等电聚焦电泳(capillary isoelectric focusing electrophoresis,cIEF)法进行分析,将样品用Nanosep超滤离心管10 000g离心干燥(约3 min)。使用去离子水重悬,吹打混匀,调整浓度至约4.0 mg/ml,取上述置换缓冲液的供试品或标准品20 μl,加入两性电解质混合物180 μl,涡旋混匀,以10 000g离心5 min。取上清液150 μl,置于进样瓶中,以7 000 r/min离心3 min。分析条件:预聚焦电压1.5 kV,持续1 min;聚焦电压3 kV,持续时间为10 min。
1.9 糖基化分析
使用超高效液相色谱法进行分析,将相当于200 μg单抗的样品溶液用50 mmol/L碳酸氢铵缓冲液稀释至2 mg/ml,取80 μl加入预处理后的Zeba Spin脱盐柱,1 500g离心2 min,收集离心液。取置换缓冲液的样品7.5 μl(即约15 μg糖蛋白),加水15.3 μl。加入配制的5% RapiGes溶液6 μl,110 ℃孵育5 min,室温冷却5 min。变性后样品加入N糖苷酶F 1.2 μl,57 ℃孵育5 min,室温冷却3 min;取释放N-聚糖的样品加入配制的RapiFluor-M标记溶液12 μl,室温孵育约5 min。加入乙腈358 μl,稀释;取HILIC μElution平板,依次取水、水/乙腈(15/85)以200 μl/孔平衡树脂。加入处理好的供试品,取甲酸/水/乙腈溶液(1/9/90)以600 μl/孔洗板,重复2次。后加入SPE洗脱缓冲液30 μl洗脱,重复3次。最后加入样品稀释剂(DMF/ACN 32/68,V/V)310 μl,即得纯化N-糖样品。上样分析。色谱条件:流动相A为50 mmol/L甲酸铵,pH 4.4;流动相B为乙腈。使用Waters H-Class超高效液相色谱仪,荧光检测器进行分析,检测器激发波长265 nm,发射波长425 nm,色谱柱为Waters Acquity UPLC Glycan BEH amide 1.7 μm,150 mm×2.1 mm;进样体积为10 μl,柱温60 ℃,样品盘温度8 ℃。采用面积归一化计算单个聚糖的含量。
2 结果
2.1 抗GD2单抗参比品的生物学活性分析结果
ADCP和CDC效应测定所得数据均符合四参数方程式:Y=(A-D)/[1+(X/C)B]+D,即在半对数坐标上呈现典型的S型曲线(见图1和图2)。在ADCP效应评价中,抗GD2单抗参比品的EC50值为(24.24±0.57)ng/ml,6次实验的RSD值为13.88%。在CDC效应评价中,抗GD2单抗参比品的EC50值为(0.49±0.003)ng/ml,6次实验的RSD值为0.61%。

图1 抗GD2单抗ADCP效应的剂量反应曲线Figure 1 Dose-response curves of ADCP effect of anti-GD2 mAb
2.2 抗GD2单抗参比品的结合活性分析结果
GD2和CD16结合活性检测所得数据也均符合四参数方程式:Y=(A-D)/[1+(X/C)B]+D,即在半对数坐标上呈现典型的S型曲线(见图3和图4)。在GD2结合活性评价中,抗GD2单抗参比品的EC50值为(17.53±2.14)ng/ml,6次实验的RSD值为1.21%。在CD16结合活性评价中,抗GD2单抗参比品的EC50值为(99.40±1.43)nmol/L,6次实验的RSD值为1.44%。

图2 抗GD2单抗CDC作用的剂量反应曲线Figure 2 Dose-response curves of CDC effect of anti-GD2 mAb
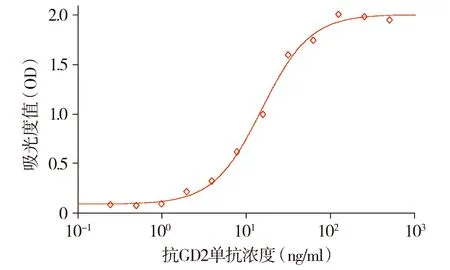
图3 抗GD2单抗的GD2结合活性的剂量反应曲线Figure 3 Dose-response curve of GD2-binding activity of anti-GD2 mAb
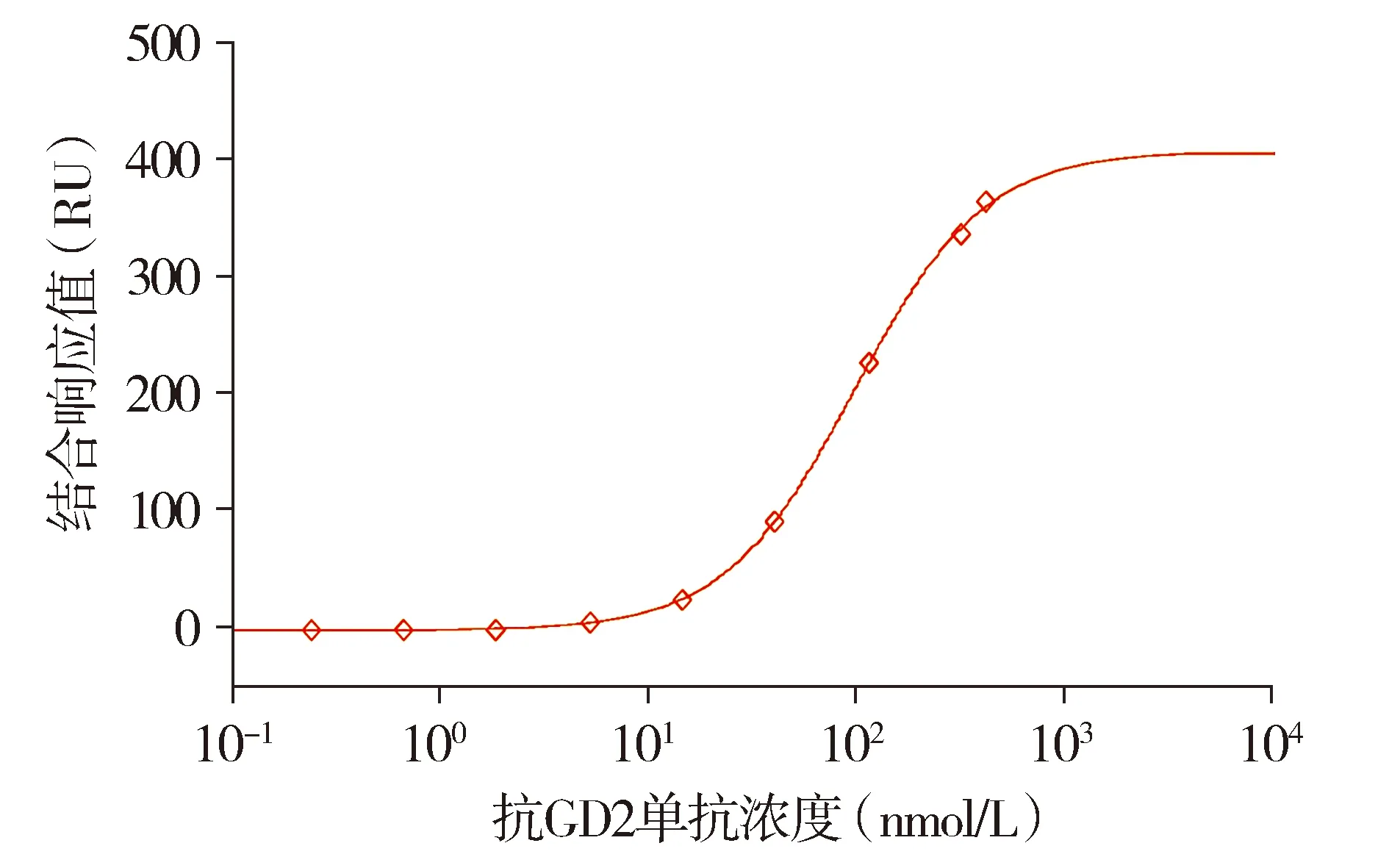
图4 抗GD2单抗的CD16结合活性的剂量反应曲线Figure 4 Dose-response curve of CD16-binding activity of anti-GD2 mAb
2.3 抗GD2单抗参比品的纯度分析结果
非还原CE-SDS分析表明,抗GD2单抗参比品的主峰面积百分比为(98.61±0.17)%(见图5),6次实验的RSD值为0.17%。还原CE-SDS分析表明,抗GD2单抗参比品的重链(HC)和轻链(LC)的峰面积百分比之和为(97.39±0.15)%(见图6),6次实验的RSD值为0.15%。SE-HPLC分析表明,抗GD2单抗参比品主峰峰面积百分比为(98.32±0.01)%(见图7),6次实验的RSD值为0.01%。
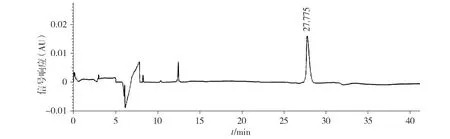
图5 抗GD2单抗纯度的非还原CE-SDS纯度分析图谱Figure 5 The non-reduced CE-SDS purity analysis of anti-GD2 mAb
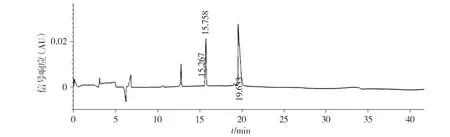
图6 抗GD2单抗纯度的还原CE-SDS纯度分析图谱Figure 6 The reduced CE-SDS purity analysis of anti-GD2 mAb
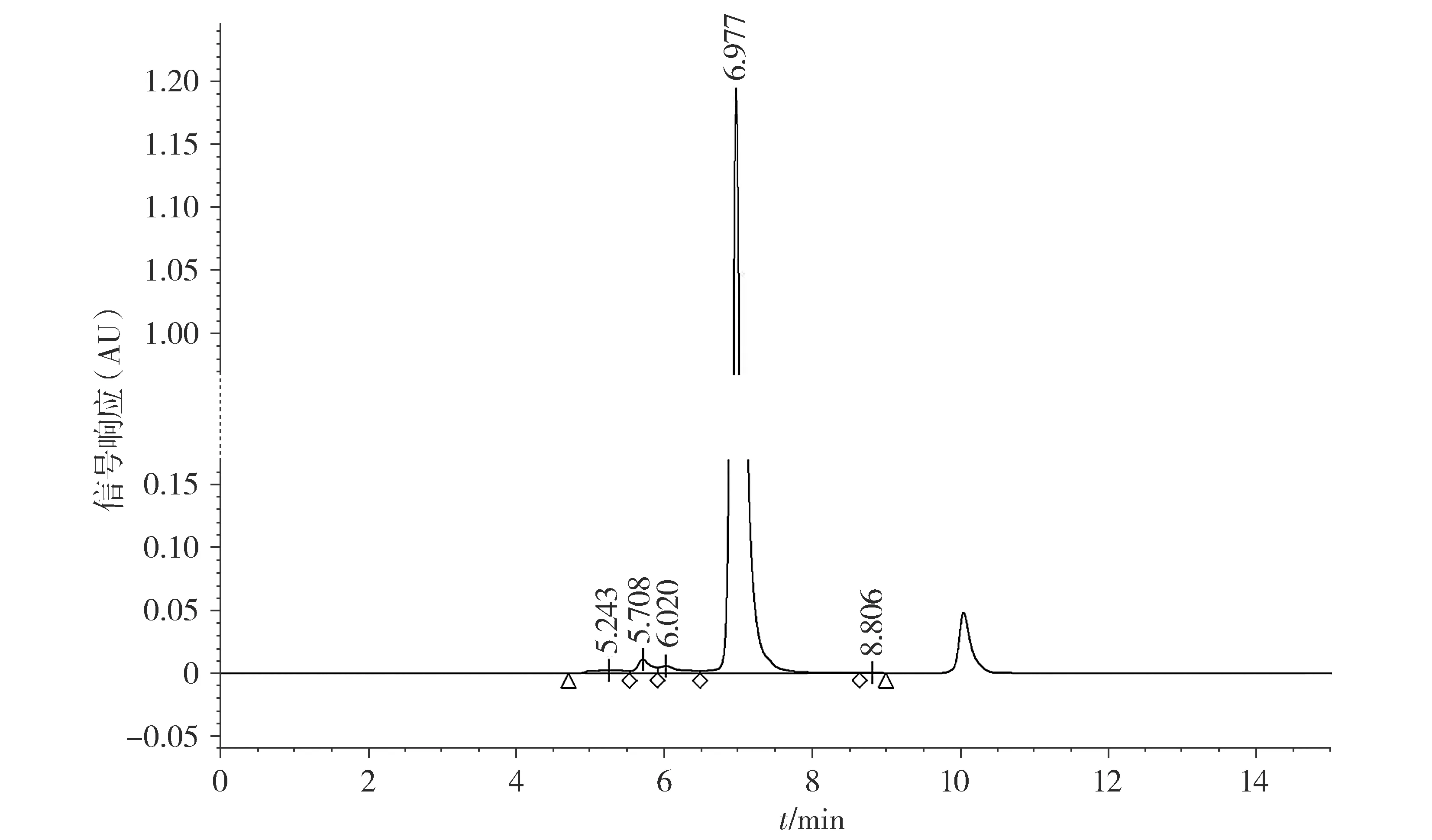
图7 抗GD2单抗纯度的SE-HPLC纯度分析图谱Figure 7 The SE-HPLC purity analysis of anti-GD2 mAb
2.4 抗GD2单抗参比品的电荷异质性分析结果
cIEF分析表明,抗GD2单抗参比品中峰“l”的峰面积百分比为(14.88±0.15)%(见图8),6次实验的RSD值为1.01%;峰“m”的峰面积百分比为(37.18±0.37)%,6次实验的RSD值为1.00%;峰“n”的峰面积百分比为(38.67±0.50)%,6次实验的RSD值为1.29%。
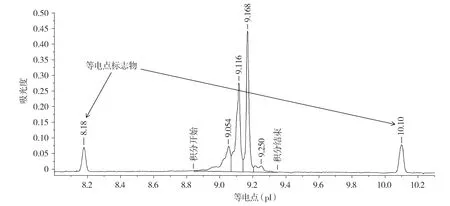
图8 抗GD2单抗的等电点及电荷异质性的cIEF分析Figure 8 The cIEF analysis of isoelectric point and charge heterogeneity of anti-GD2 mAb
2.5 抗GD2单抗参比品的糖基化分析结果
每个单糖所占比例由荧光图计算所得,结果见图9。将相对百分比≥1%的单糖按照岩藻糖基化和半乳糖基化分为两类进行统计分析,结果显示抗GD2单抗岩藻糖基化单糖合计所占比例为(97.41±0.04)%,6次实验的RSD值为0.04%,半乳糖基化单糖合计所占比例为(18.93±0.07)%,6次实验的RSD值为0.37%。
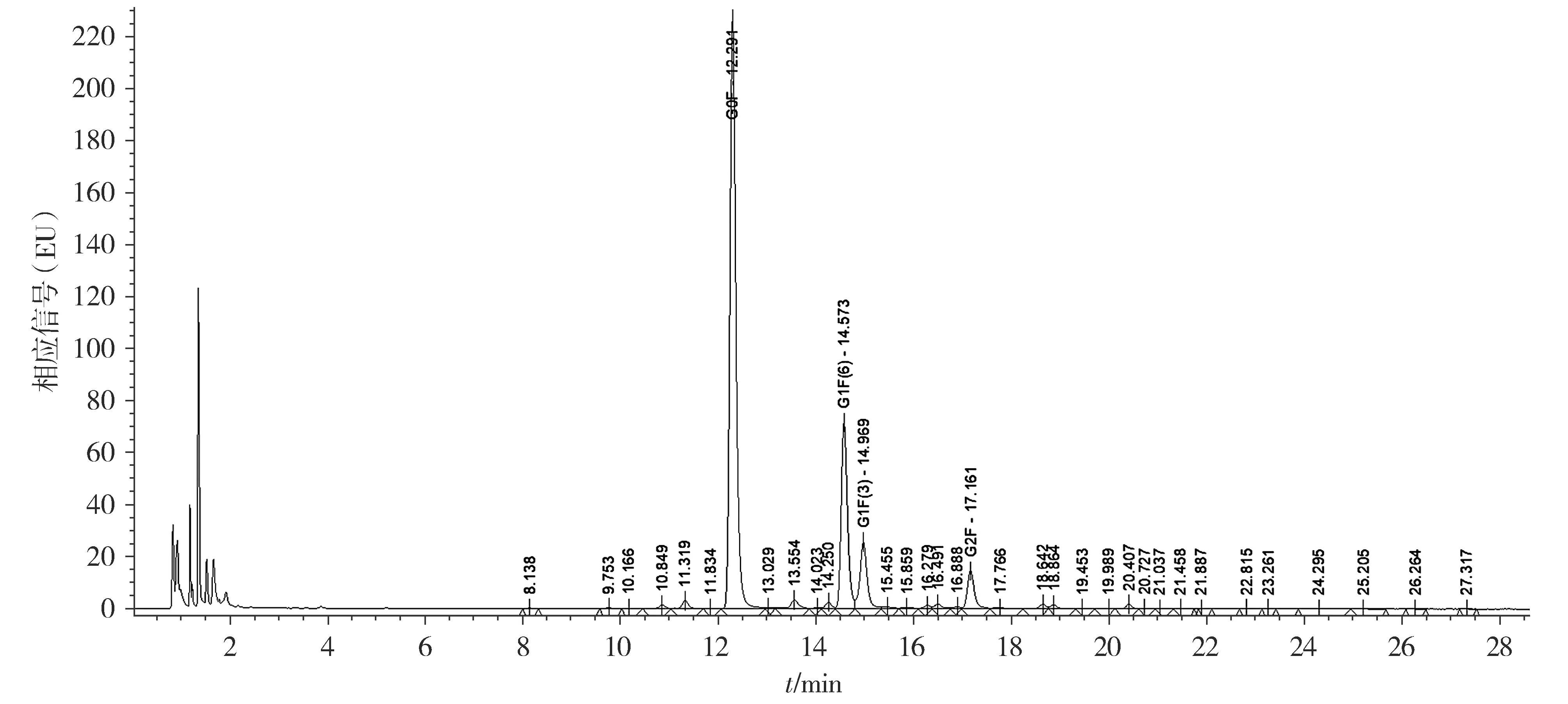
G0F、G1F(6)、G1F(6)、G2F中的G表示半乳糖,F表示岩藻糖,G后面的数字表示连接到两个N乙酰氨基葡萄糖(GlcNAc)的末端半乳糖(galactose)的数量,F后面括号中的数字表示岩藻糖的数目(没有数字表示仅存在核心岩藻糖)图9 抗GD2单抗的糖基化分析Figure 9 Glycosylation profiling of anti-GD2 mAb
3 讨论
《中国药典》2020年版明确提出应采用现有先进的分析手段,从物理化学、免疫学、生物学等角度对单抗制品进行全面的分析,并提供尽可能详尽的信息,以反映目标产品内在的天然质量属性。其中最为关键的一项是应依据单抗预期的、潜在的作用机制或工作模式(可能不限于一种)建立相应的生物学分析方法[10]。以往研究表明,抗GD2单抗的作用机制包括Fc段介导的ADCC、ADCP和CDC效应,以及Fab段介导的直接杀伤作用和抑制循环中肿瘤细胞与细胞外基质蛋白成分的结合和细胞归巢等[12,13]。据此,本研究从Fc段和Fab段两个层面对抗GD2抗体的生物学活性进行了综合全面的评价。包括以基于细胞水平杀伤的ADCP和CDC效应实验和基于SPR-Biacore的CD16结合实验(代替细胞水平的ADCC效应实验)来评估抗GD2抗体的Fc段生物学功能,以基于ELISA的GD2结合实验(代替细胞水平的直接杀伤实验)来评估抗GD2抗体的Fab段生物学功能。由于细胞水平的生物学功能研究更能有效地模拟抗体的真实功能[14],因此本研究下一步将建立基于细胞水平的ADCC和Fab段生物学功能实验,并与现有的结合实验进行平行性比较。但在常规的质量控制活动中,应根据需要选择既能反映真实生物学活性又便于实施操作的实验手段。
抗体药物的纯度检测和控制贯穿于工艺开发的整个生命周期中。SDS-聚丙烯酰胺毛细管电泳(CE-SDS)和分子排阻液相色谱(SE-HPLC)是两种主要的抗体药物纯度分析方法[15]。由于这两种方法检测原理的不同,导致其反映的抗体纯度的侧重点也不同。比如,非还原型CE-SDS侧重将完整抗体和不完整抗体(非共价键连接的片段)分开,还原型CE-SDS能将非糖基化重链和断裂片段检测出来,而SE-HPLC则是在比较温和的实验条件下将聚体(共价和非共价结合)和单体有效分离。本研究中同样将两种方法共同作为抗GD2抗体纯度的质控方法。抗GD2抗体部分生物学活性的发挥依靠Fc段功能,而重链的糖基化与Fc段功能有关,因此理论上在还原型CE-SDS中应该增加对非糖基化重链的控制。但是考虑到其活性已经从ADCP、CDC以及GD2和CD16结合等4个方面进行了全面的质控,故未在还原型CE-SDS中纳入非糖基化重链的控制。
由于抗体是一种具有复杂翻译后修饰的生物大分子,决定了抗体药物的微观不均一性。而几乎所有这种微观不均一的变体均能导致抗体表面电荷分布的差异,因此电荷的变化也成为监测抗体降解和生产工艺一致性分析的重要指标。目前用于电荷异质性研究的常用方法有IEF、cIEF、icIEF、CEX、RP-HPLC、LC-MS等,每种方法各有优缺点。本研究使用的cIEF可根据抗体净电荷的变化精确地分离鉴别每种电荷变体,并选择了占比前三位的电荷变体建立了质控方法。
抗体的糖基化是一种复杂的翻译后修饰,对单抗药物的疗效、稳定性、免疫原性、药代动力学性质等具有重要影响[16,17]。例如,核心岩藻糖基化的缺失会由于对FcγR受体的亲和力增加而增强ADCC和ADCP效应,末端半乳糖基化减少会降低CDC效应等。有研究表明,在GnT1缺陷型CHO细胞中表达的抗GD2抗体携带Man5-GlcNAc2糖基化,而α1,6-岩藻糖缺失,与亲本抗GD2抗体或其他Fc增强突变体相比,在体外和体内抗肿瘤方面,作为治疗性抗体的潜力更大[18]。另外,虽然半乳糖基化对抗体效应功能的影响目前了解得相对较少,但是有研究表明半乳糖基化是以CDC为作用机制的单抗的关键质量属性,并且深入分析了末端半乳糖与单抗治疗中补体激活的结构-功能关系[19]。基于此,本研究选择了通过荧光检测器的检测确定各个糖型的比例,并通过岩藻糖基化和半乳糖基化的抗体比例建立了糖基化检测的质控方法。
综上所述,本研究根据现有技术并依据《中国药典》和ICH Q6B的指导原则[10,11],针对抗GD2单抗的质量控制开展了研究。根据抗GD2单抗产品的特性和作用机制,确定了关键质量属性并建立了基于不同原理的质控方法,对我国抗GD2单抗药物的研发具有借鉴意义。随着新技术的出现以及对该靶点作用机制更深入的研究,其质控方法也将会随之不断发生变化。
猜你喜欢
中国临床新医学(2022年8期)2022-09-08
中国典型病例大全(2022年7期)2022-04-22
健康之家(2021年2期)2021-07-01
医学食疗与健康(2021年27期)2021-05-13
证券市场红周刊(2019年6期)2019-06-11
科学导报(2019年5期)2019-06-11
祝您健康(2018年12期)2018-11-27
食品界(2018年8期)2018-09-03
分析化学(2018年12期)2018-01-22
中学生数理化·高二版(2016年6期)2016-05-14
