
Genotypic and phenotypic spectra of NBEA-related neurodevelopmental disorder with epilepsy: a case series and literature review
2022-11-08 08:36:38ZouPanChenChenFeiYinJingPeng
World Journal of Pediatrics 2022年9期
Zou Pan · Chen Chen · Fei Yin · Jing Peng ,2
NBEA
(MIM # 604,889) is a novel disease causative gene that responds for neurodevelopment disorder with or without generalized epilepsy (NEDEGE, MIM #619,157). It encodes neurobeachin protein, a multi-domain neuro-specif ic scaffolding protein that plays a vital role in vesicle traffi cking and synaptic structure.NBEA
was initially observed in an idiopathic autism patient, and thusNBEA
has been regarded as a candidate autism gene for nearly two decades [ 1— 4]. In 2018, Mulhern et al. reported 24 individuals with de novo heterozygousNBEA
variants with neurodevelopmental delay with autism and early-onset generalized epilepsy, which led the Online Mendian Inheritance in Man (OMIM) team to identify it as a disease-causing gene for NEDEGE [ 5]. Since then, only a fewNBEA
-related cases with similar clinical phenotypes have been reported [ 6— 8].Herein, we reported three cases with NEDEGE and identif ied three novelNBEA
variants. Furthermore, we systematically reviewed the patients withNBEA
variants and summarized the phenotypic and genotypic features of theNBEA
-related disorder.The clinical data, including demographics, clinical features, auxiliary examination, and neuroimaging, were collected retrospectively from three patients who were admitted in the Department of Pediatrics, Xiangya Hospital of Central South University because of epileptic encephalopathy of unknown etiology. Brief summary is shown in Table 1.Trio whole-exome sequencing was subsequently performed,and variants were validated by Sanger sequencing. The pathogenic assessment of candidate variants was implemented according to the 2015 American College of Medical Genetics and Genomics (ACMG) standard guidelines[ 9]. This study was approved by the Ethics Committee of Xiangya Hospital of Central South University, China (#201,603,205). Informed consent was obtained from the guardians of all the children.
We have performed a systematic literature review by searching the online databases, including PubMed, Wanfang,and CNKI as of October 2021, using the keywords “NBEA
”,“neurobeachin”, “epilepsy” and “intellectual disability”.All publications were reviewed, and the clinical and genetic information of the individuals in each study were summarized. All the variants were recoded according to theNBEA
reference transcript NM_015678.4 (NP_056493.3). The phenotypic and genotypic features of previously reported and newly identif ied individuals withNBEA
-related disorders were systematically reviewed.Patient 1, a 59-month-old girl, had a focal seizure with fever at the age of 16 months. Levetiracetam (LEV) was prescribed with no effi cacy. Sodium valproate (VPA) led to a one-year, seizure-free period. Unfortunately, her seizures relapsed at the age of 28 months, accompanied by the twitching of the left side of her mouth and the blinking of the right eye. Topiramate (TPM) and lamotrigine (LTG) were administrated but ineff ective. Finally, clobazam (CLB) was added to the treatment and became seizure-free. She had cognitive impairment and stereotyped behaviors. At the last follow-up visit at the age of 59 months, she could walk but could not speak and communicate with others. The brain MRI showed enlarged bilateral lateral ventricles and temporal horns. The video electroencephalogram (VEEG) revealed slowed posterior rhythm and abundant epileptic discharges in the left temporal-parietal-occipital region (Supplemental Fig. 1). In addition to epilepsy, she also suff ered from recurrent infection and thrombopenia. Platelet tests showed lower counts (ranging from 78 to 110 × 10 9 /L) and volumes (mean platelet volumesranged from 6.9 to 7.5 fL). The patient had no physiological abnormalities, and metabolic screenings were normal. Whole exome sequencing was performed, and a de novo missense variant ofNBEA
(c.7948C > G, p.P2650A) was detected. This variant was not observed in gnomAD database and was predicted to be deleterious and highly conserved. The REVEL rank score and Combined Annotation Dependent Depletion(CADD) phred score were 0.72 and 23.6, respectively. Thus,we regarded it as a pathogenic variant (PS2 + PM2 + PP3).Table 1 Clinical features of the three patients with a variant
anti-seizure medications, clobazam, electroencephalograph, global developmental delay, ketogenic diet, topiramate, sodium valproate, head circumference, height. † Physical examination was performed at 3 y 11 mon for individual 1, 4 y 7 mon for individual 2, and 3 y 5 mon for individual 3
Variables Patient No. 1 Patient No. 2 Patient No. 3 Gender Female Male Male Age of the last follow-up 4 y 11 mon 5 y 5 mon 4 y 2 mon Age of seizure onset 1 y 4 mon 2 y 5 mon 3 y 4 mon Seizure types Focal motor, atypical absence, febrile sensitive Tonic, Tonic—clonic, Myoclonic, Epileptic Spasms, Atypical absence Tonic—clonic, Myoclonic Eff ective ASMs CLB CLB, KD TPM, CLB Neuropsychiatric disorder Severe GDD, Autism-like behavior Severe GDD Mild GDD Pysical examination † Normal Normal HC: 43 cm, H: 94 cm EEG patterns Background: posterior rhythm slow down;Interictal: bilateral un-synchronously discharge, left temporal-parietal-occipital region predominantly MRI features Mild enlargement of bilateral lateral ventricles and temporal horns Background: slow down;Interictal: generalized multifocal spike and slow waves Background: diff use slow waves;Interictal: generalized 1.2—2.5 Hz spike-slow wave Normal Normal Variants NM_015678.4:c.7948C > G, p.P2650A NM_015678.4: c. 3949C > T:p.R1317* NM_015678.4:c.5649C > A,p.S1883R
Patient 2 is a 65-month-old boy. He presented with absence seizures at the age of 29 months, and he developed tonic—clonic seizures after 2 weeks. No anti-seizures medication was administered until the age of 53 months when his seizures became more frequent and severe. The brain MRI was normal, and the VEEG showed slow background rhythm, generalized spike and slow-wave discharges and myoclonic seizures (Supplemental Fig. 1 B1-4). TPM and VPA were prescribed but without efficacy. Myoclonictonic and myoclonic seizures during sleep were captured by VEEG (Supplemental Fig. 1 B5-8). He was then treated with CLB, LEV and ketogenic diet, and seizure episode was reduced to 1 time per month. Gesell assessment showed a severe global developmental delay. However, global development was gradually improved as the seizures were decreased. At the last follow-up visit at the age of 65 months,he could communicate with other people and could attend school normally. In addition, the physical inspection and routine auxiliary tests were normal, but a de novo nonsense variant of theNBEA
gene (c.3949C > T, p.R1317*) was detected by exome sequencing. This nonsense variant was not present in gnomAD database. Moreover, it caused a lossof-function event forNBEA
which was a haploinsuffi cient sensitive gene. We have classif ied it to be a pathogenic variant (PVS1 + PS2 + PM2).Patient 3, a 50-month-old boy, had first generalized tonic—clonic seizure at 40 months of age. He received VPA and methyl-prednisolone pulse therapy but without response.He was then referred to our hospital. His brain MRI was normal, and VEEG showed diff use slow waves with generalized 1.2—2.5 Hz spike-slow waves. Generalized tonic—clonic seizures and myoclonic seizures were detected as well (Supplemental Fig. 1 C1—7). Then TPM, CLB, and another methylprednisolone pulse therapy were given. Fortunately, seizures and epileptic discharges disappeared, and his psychomotor development was improved. At the last follow-up, he had become seizure-free for six months, and VEEG showed no epileptic discharge (Supplemental Fig. 1 C8). The Griffi th assessment showed mild global development delay, but the autism scanning results were normal. His physical examination revealed microcephaly (43 cm, < -3SD) and mild short stature (94 cm, -2SD—1SD). Exome sequencing detected a de novo missense variant of theNBEA
gene (c.5649C > A,p.S1883R), which was not recorded in population database.The REVEL rank score and CADD phred score were 0.51 and 29, respectively. In addition, the scSNV database predicted that his variant might aff ect mRNA splicing. We regarded it as a potential pathogenic variant (PS2 + PM2 + PP3).Thirty-f ive individuals withNBEA
variants were identif ied during our literature search, and systematically reviewed in this study Table 2), with no strong gender bias (male:female = 18:17).Twenty-two of them were reported to have variable seizures with the disease onset ranging from 8 months to 19 years of age.The majority (91%) of epileptic patients suff ered from generalized epilepsy, and tonic—clonic seizure (59%) was the most common seizure type. Other seizure types included myoclonic(41%), typical/atypical absence (32%) and tonic (23%). Status epilepticus also occurred in a few patients. Seizures were usually intractable, and only nine of 22 patients became seizure-free.68% (male, 13; female, 10) of individuals suff ered from mild to severe intellectual disability, and most were moderate. Language delay was also common; 78% (n
= 25) of individuals had language delay and four patients were completely non-verbal.About half of individuals had autism or autism-like behavioral problems, and males (n
= 13) were more likely to have autism features than females (n
= 6).Table 2 Phenotypic and genotypic information of patients with a variant
References This study This study This study[ 7][ 8][ 5][ 5][ 5][ 5]Autism++++++Language delay LD LD LD LD LD LD NV LD LD GDD Severe Severe Mild Mild Moderate Severe Moderate Eff ective ASMs CLB CLB, KD TPM, CLB KD KD, VPA,ESM Seizure+++++++++++Mutation types Seizures freedom Missense Nonsense Missense Missense Frameshift Nonsense Missense Frameshift Variants c.7948C > G;p.Pro2650Ala c.3949C > T; p.Arg1317*c.5649C > A;p.Ser1883Arg c.5899G > A;p.Gly1967Arg c.5258_5279del;p.Ala1753Valfs*13 c.1006C > T; p.Arg336* Nonsense c.6829C > T;p.Arg2277*c.3994C > T;p.Pro1332Ser[ 5][ 5][ 5][ 5][ 5][ 5][ 5][ 5][ 5][ 5][ 5]+++LD LD LD LD NV LD LD LD Mild Moderate Moderate Mild Moderate Moderate Moderate Moderate VPA, ESM LTG VPA, LEV VPA, LEV,TPM++++++++++Nonsense Frameshift Splicing site Nonsense Nonsense Frameshift Frameshift Missense Nonsense Missense Nonsense c.6313G > T;p.Glu2105*c.4484del;p.Asn1495Ilefs*17 Gender Female Male Male Female Male Male Female Male Male Case ( n)1 2 3 4 5 6 7 8 9 c.7294_7295dup;p.Glu2433Argfs*3 c.7707 + 2 T > C c.6868C > T;p.Gln2290*c.7462G > T;p.Glu2488*c.7230del;p.Asp2411Ilefs*21 c.3183delA;p.Glu1062Argfs*8 c.1448C > T;p.Ala483Val c.6637C > T;p.Arg2213*c.2836C > T;p.His946Tyr c.3832C > T;p.Arg1278*Male Female Female Male Female Male Male Female Female Female Female 10 11 1213 14 15 16 17 18 19 20
Table 2 (continued)
anti-seizure medications, clobazam, ethosuximide, diazepam, ketogenic diet, levetiracetam, lamotrigine, language delay, non-verbal, topiramate, unkown, sodium valproate
References[ 5][ 5][ 5][ 5][ 5][ 5][ 5][ 5][ 5][ 6][ 10][ 1][ 3][ 4][ 2]Autism+++++++++Language delay LD LD LD LD LD NV LD NV GDD Moderate Moderate UK Moderate UK Mild UK Mild Eff ective ASMs LEV VPA, LTG DZP, LEV,VPA, CZP Seizure++++++++++Mutation types Seizures freedom Frameshift Missense Nonsense Multigene deletion Intragenic deletion Intragenic deletion Intragenic deletion Intragenic deletion Intragenic deletion Multigene deletion Intragenic deletion Multigene deletion Multigene deletion Multigene deletion Balanced chromosomal translocation Variants c.4715C > A;p.Ser1572*chr13:33,957,317—36,828,237 × 1 c.3362del;p.Asn1121Metfs*9 c.8401G > A;p.Glu2801Lys chr13:35,590,335—35,940,429 × 1 chr13:35,574,513—36,163,037 × 1 chr13:36,038,249—36,141,224 × 1 chr13:35,963,197—36,125,577 × 1 chr13:35,700,830—35,887,000 × 1 chr13:35,940,229—39,099,628 × 1 chr13:35,750,001—35,785,000 × 1 del(13)(q12q13)del(13)(q13.2q14.1)del(13)(q12.3q14.3)t(5;13)(q12.1;q13.2)Gender Female Male Female Male Male Male Male Male Female Female Female Male Male Female Male Case ( n)21 22 23 24 25 26 27 28 29 30 31 32 33 34 35
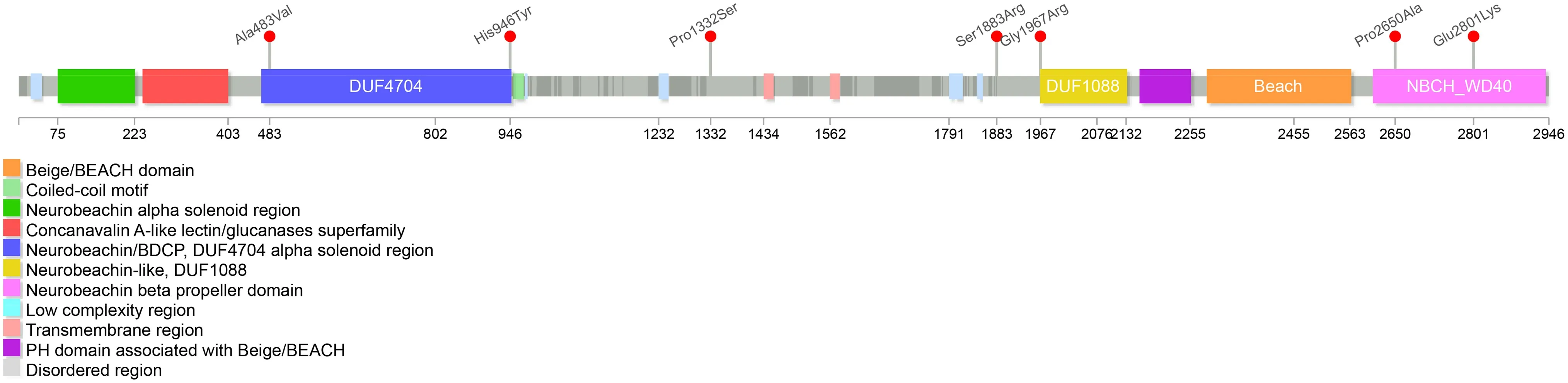
Fig. 1 Missense variants of NBEA gene carried in patients. Seven pathogenic missense variants are detected in patients, three located at the WD40 domain, two at the DUF4704 domain, one located at the DU1088 domain and one near the DUF1088 domain
Thirty-two differentNBEA
variants from separate families were reported [ 1— 8, 10], and three novel variants(p.P2650A, p.R1317*, and p.S1883R) were identif ied from the three unrelated patients in our cohort (Table S1). All of the variants were de novo, except one due to unavailability for familial validation. The majority of single nucleotide variants (SNV) were null, including 9 nonsense variants, 6 frameshift variants and 1 splicing site variant, and the other 12 were structure variants. Only 7 missense variants were reported (Fig. 1). Three were located at the WD40 domain,two at the DUF4704 domain, one located at the DU1088 domain and one nearby at the DUF1088 domain. There were no hot positions or domains. We have performed genotype—phenotype relationship analyses and found no signif icant diff erence between null SNV and missense variants. However, patients with structure variants had a lower incidence of developmental delay and seizures (Table S2).Neurobeachin, a neuron-specif ic scaff old protein encoded by theNBEA
gene, is a large (327 kDa) multi-domain protein with at least seven distinct motifs/domains [ 11]. TheNBEA
is highly expressed in the brain and plays an important role in vesicle traffi cking, dendritic spines formation, synaptogenesis and synaptic transmission [ 12, 13]. This gene has been found to be a haploinsuffi cient sensitive gene, andnbea
( +/-) mice have been found to present with an autism spectrum disorder that in vivo studies have suggested was caused byNBEA
’s eff ect on synaptic plasticity [ 14]. For this and other reasons,NBEA
was considered an autism candidate gene [ 1— 4]. With moreNBEA
variants detected in patients with neurodevelopmental delay and epilepsy, the association between theNBEA
gene and NEDEGE has become clearer [ 5]. Herein, we reported cases of the three unrelated Han Chinese epileptic encephalopathy patients with a novelNBEA
variant.The epileptic pattern ofNBEA
has been characterized by generalized epilepsy with myoclonic seizures [ 5]. According to our literature review, most individuals had tonic—clonic seizures, followed by myoclonic epilepsy. No specif ic EEG features could be summarized from the current cohort. However, slow-wave on background and generalized epileptic discharge were common, andNBEA
-caused epilepsy seemed to be intractable. Less than a third of patients became seizure-free, and most of them accepted two or more ASMs.LEV, VPA, LTG and ketogenic diet might be effective according to the published data[ 8].Cognitive and neuropsychiatric disorders are common comorbidities among intractable patients [ 15]. Our patients showed dramatic regression of motor and psychodevelopmental levels after seizure onset. Interestingly,with their seizures controlled, cognitive and motor development were evidently improved, suggesting that mental retardation was not just the result of neurobeachin abnormalities, but was also aff ected by the severity of epileptic discharge. In addition, patient 1 suff ered from thrombocytopenia. Studies have shown that neurobeachin has participated in cytoskeleton formation of platelets and modulates the platelets secretion [ 16, 17]. However, the association between thrombocytopenia and remains obscure.
No obvious diff erences in epileptic or neurodevelopmental phenotypes have been observed between null SNV and missense variants. Interestingly, language delay and seizures were less common among structural variants, which might be attributed to prior studies focusing on autism spectrum disorders. Six of the seven missense variants caused seizures.Two were located at the C-terminal WD40 presenting with focal and febrile-sensitive seizures, and one was located at the DUF1088 domain. Both DUF1088 and WD40 might be implicated in the SAP102 binding of PH-BEACH and involved in AMPA- and NMDA-type receptor traffi cking [ 12]. A C.elegans model showed that the missense variant in DUF1088 destroyed the traffi cking of potassium channels in neurons [ 7].This indicated that pathogenic variants within the DUF1088-PH-BEACH-WD40 domain might aff ect the traffi cking and synaptic targeting ion channels or neurotransmitter receptors,and disrupt the regulation of neuron excitability, thus causing epilepsy [ 18].
In conclusion, we have identif ied three novel pathogenic variants of theNBEA
gene among the Chinese Han cohorts; and we summarized the phenotype and genotype features of patients found in our systematric literature review. Neurodevelopmental delay was the most signif icant feature, followed by seizures, and autism behavior.Seizure control will benef it for improving development.Supplementary Information The online version contains supplementary material available at https:// doi. org/ 10. 1007/ s12519- 022- 00567-9.
Acknowledgements
Thanks a lot for patients and their parents’support.Author contributions
ZP: data curation, formal analysis, methodology, writing—original draft, writing—review & editing; CC: data curation, resources, funding acquisition, writing—review & editing;FY: resources; JP: resources, supervision, project administration, writing—review & editing.Funding
This study was funded in part by the National Natural Science Foundation of China (Grant no. 81771409 and 82071462) and Natural Science Foundation of Hunan Province (Grant no. 2021JJ40969).Data availability
statement The data presented in the study are included in the article/supplementary materials, and further inquiries can be directed to the corresponding author.Declarations
Conflict of interest
No f inancial or non-f inancial benef its have been received or will be received from any party related directly or indirectly to the subject of this article.Ethical approval
This study was approved by the Ethic s Committee of Xiangya Hospital of Central South University, China (human study/protocol # 201603205). Informed consents were obtained from the parents of all subjects. World Journal of Pediatrics2022年9期
World Journal of Pediatrics2022年9期
- World Journal of Pediatrics的其它文章
- Association between genetic polymorphisms of base excision repair pathway and glioma susceptibility in Chinese children
- Evaluation of proximal tubule functions in children with COVID-19:a prospective analytical study
- Using echocardiography in newborn screening for congenital heart disease may reduce missed diagnoses
- Impact of comorbidities on the prognosis of pediatric vasovagal syncope
- Eff ect of early feeding practices and eating behaviors on body composition in primary school children
- Asthma mortality among children and adolescents in China,2008–2018