
巨噬细胞极化在痛风性关节炎中的研究进展
2022-11-05 05:14郝佳瑶黄逸晨马宁宁沈海丽
医学研究杂志 2022年10期
郝佳瑶 黄逸晨 张 娟 马宁宁 沈海丽
痛风性关节炎(gouty arthritis,GA)属于风湿性疾病中晶体关节炎的一种,是由关节和关节周围组织中沉积的尿酸单钠盐(monosodium urate,MSU)晶体引起的急性或慢性炎性反应。 在痛风性关节炎发病机制中,作为固有免疫的巨噬细胞(macrophages,Mϕ)发挥着重要角色[1]。 在受到不同微环境刺激后,Mϕ极化为M1 型和M2 型,并分泌多种细胞因子。 M1 型极化发生在炎症的初始阶段,而M2 型极化在炎症缓解过程中占主导地位。 Mϕ 极化状态之间的转换会导致慢性炎症、代谢紊乱,甚至发生自身免疫疾病[2]。 痛风性关节炎的发病及缓解机制目前仍在探究中,而Mϕ 极化可能发挥着重要作用,现对近年来痛风性关节炎中巨噬细胞极化类型、巨噬细胞极化方式、主要的信号通路以及治疗靶点等相关研究进行归纳总结,以期为后续深入研究痛风性关节炎的发病机制及治疗靶点提供新的思路。
一、巨噬细胞极化类型
Mϕ 极化是指受到细胞因子和微环境的影响后进一步分化为M1 型和M2 型,如图1 所示。 M1 型即经典型活化Mϕ,其表型包括趋化因子2、CD86、缺氧诱导因子-1α(hypoxia inducible factor-1α,HIF-1α)等,由肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)、干扰素γ(interferon-γ,IFN-γ)、脂多糖(lipopolysaccharide,LPS)诱导形成,产生TNF-α、白细胞介素-1(interleukin-1,IL-1)、活性氧(reactive oxygen species, ROS)、金属蛋白酶等细胞因子,发挥促炎和吞噬作用[2]。 M2 型即选择型活化Mϕ,又可分为M2a、M2b、M2c、M2d4 个亚型,主要表达CD36、CD206 和CD163,由白细胞介素-10(interleukin-10,IL-10)、白细胞介素-4(interleukin-4,IL-4)诱导形成,分泌IL-10、转化生长因子-β(transforming growth factor-β,TGF-β)、血管内皮生长因子等,在炎症和组织修复途径中起作用,M2 型相比于M1 型,结构和功能更加多样化,M2a 型主要发挥促进细胞增殖和迁移、清除凋亡细胞的作用[3]。M2b 型具有使细胞成熟,维持稳态,合成细胞外基质的功能[4]。 M2c 型能消除炎症,合成细胞外基质,组织修复,产生生长因子[5]。 M2d 型可以促进血管再生和伤口愈合[6]。
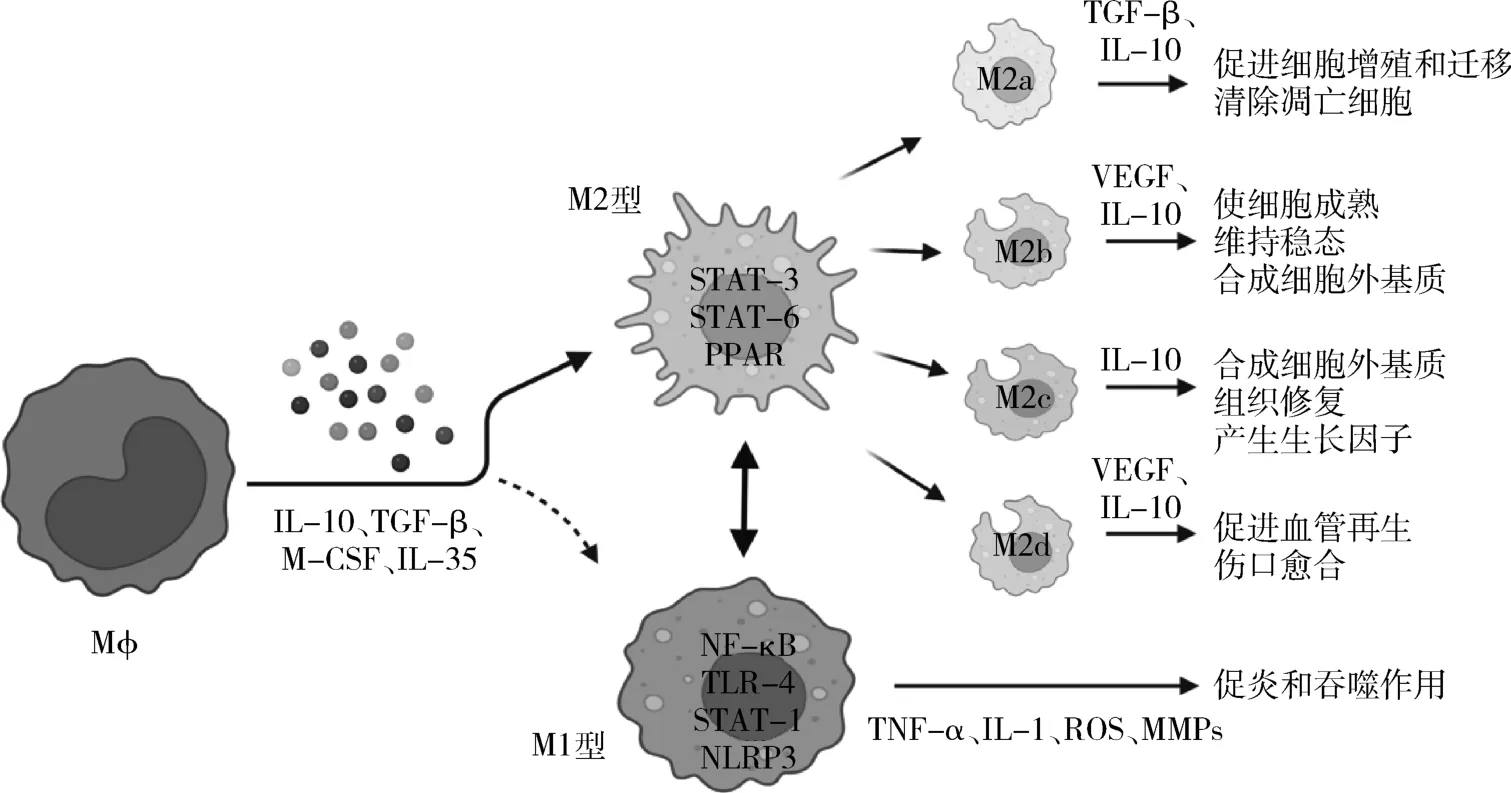
图1 不同巨噬细胞极化类型及作用
二、痛风性关节炎中巨噬细胞极化方式
1.缺氧诱导极化:缺氧是Mϕ 募集和极化的关键驱动因素。 缺氧组织会分泌高浓度的趋化因子、HIF-1 和内皮素-2,吸引Mϕ 到缺氧区域[7]。 急性缺氧环境有利于M2 型极化,慢性缺氧环境诱发M1型极化。 炎症初期(1 ~4 周)可引起急性缺氧,从第4 周开始诱发M2 型极化。 M2 型发挥抗炎和组织修复作用来减轻炎症(4 ~8 周),如果炎症得不到缓解,则长时间缺氧(>8 周)更有利于M1 型极化和生存,导致慢性炎症性疾病[8]。 在痛风性关节炎患者中已经观察到,炎症严重程度和缺氧呈正相关,炎症水平越高,HIF-1α 水平越高[9]。
2.骨髓来源巨噬细胞更具可塑性:Mϕ 的某些极化状态与巨噬细胞发育起源相关,Mϕ 不仅包括骨髓来源的循环单核细胞,还包括胚胎等其他来源。 骨髓来源的Mϕ 比胚胎Mϕ 更容易受到局部组织信号的影响和随后的极化,这种极化也称之为固有巨噬细胞极化[10]。
在痛风性关节炎中,虽然MSU 晶体均能在骨髓和原位Mϕ 诱导炎性反应,但是急性期炎症的启动更倾向于MSU 晶体诱导骨髓来源Mϕ 发生氧化应激,促进单核细胞趋化蛋白-1(monocyte chemoattractant protein-1,MCP-1)、巨噬细胞炎性蛋白-2(macrophage inflammatory protein-2,MIP-2)、趋化因子上调,促使单核细胞进入滑膜环境,参与痛风性关节炎急性发作[11]。
3.LPS 和IL-4 诱导不同表型极化:CD4+辅助性T 细胞(helper T cell,TH)分泌和释放的不同的细胞介质同样介导Mϕ 的极化,这称为外源性极化。Th1 细胞壁释放LPS,驱动Mϕ 向M1 型极化,而Th2细胞分泌IL-4,驱动M2 型极化。 IL-4 通过Stat6依赖的途径耗尽作为诱导型一氧化氮合酶(inducible nitric oxide synthase,iNOS)/一氧化氮合酶(nitric oxide synthase2,NOS2)底物的精氨酸来抑制炎性介质NO 的产生,抑制NO 的产生会导致M1 型的丧失[12]。痛风性关节炎急性期,LPS 介导的M1 型极化中,TOLL 样受体4 通过激活磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)/蛋白质丝氨酸苏氨酸激酶(protein serine threonine kinase,Akt)通路和哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)通路触发M1 型极化[13]。 在痛风性关节炎缓解期,IL-4 诱导Mϕ 释放抗炎性细胞因子TGF-β,抑制NF-κB 通路并诱导Mϕ 向M2 型极化[14]。
三、痛风性关节炎不同疾病期与巨噬细胞极化
痛风性关节炎中,MSU 晶体沉积在关节和关节周围组织,导致急性关节炎症。 常驻Mϕ 或单核细胞通过TLR2、TLR4 识别和吞噬沉积的MSU 晶体。 在痛风性关节炎急性炎症期,MSU 晶体刺激后,Mϕ 倾向于表达M1 促炎表型CD86[15]。 MSU 晶体显著诱导IL-1β 和一氧化氮合酶(nitric oxide synthase,NOS)表达,IL-1β 刺激中性粒细胞活化,释放出大量的中性粒细胞胞外诱捕网(neutrophil extracellular traps,NETs)、活性氧,趋化因子等,血液中的中性粒细胞被炎性因子、趋化因子等诱导至关节间隙,引发剧烈炎性反应[16]。
在痛风性关节炎自行缓解期, Mϕ 受MSU 晶体刺激后产生TGF-β,抑制NF-κB、NLRP3 炎性小体通路,减少促炎性细胞因子IL-1β 分泌,增加抗炎性细胞因子IL-10 的表达,进一步诱导精氨酸-1 的表达,促使Mϕ 向M2 型中M2c 为主导极化[17]。MSU 还可以刺激中性粒细胞聚集,产生NETs 诱捕MSU 晶体,并通过自噬途径分解MSU 晶体而缓解炎症[18]。 上述研究表明,痛风性关节炎急性期,MSU 晶体诱导Mϕ 向M1 型极化,释放促炎介质,而当急性炎症进展到一定程度,MSU 晶体会进一步诱导Mϕ 向M2型极化,限制炎症的发展并诱导炎症的消退,这可能是痛风性关节炎具有“自限性”的原因之一[19]。
四、痛风性关节炎中巨噬细胞极化相关信号通路
1.NF-κB 信号通路:NF-κB 是典型的促炎信号通路,在炎症状态下,IL-1β、TNF-α、IL-6 和iNOS 等促炎介质主要受转录因子NF-κB 的调节[20]。核转录因子κB 抑制蛋白α(inhibitor of NF-κB, I-κBα)的磷酸化、泛素化和降解是NF-κB 活化的必要前提。 MSU 晶体被认为是促进I-κBα 磷酸化和NF-κB 激活的最有效的促炎刺激因子之一,可促进GA 患者和Mϕ 产生大量促炎性细胞因子[21]。 NF-κB 通路同样是Mϕ 极化反应的调节因子,许多M1 基因的启动子区域都含有NF-κB 结合位点,包括IL-6、iNOS 和MCP-1,此外,p50/NF-κB 同源二聚体被发现能够协调M2 型极化并抑制M1 细胞因子的表达,痛风性关节炎大鼠模型的滑膜组织中发现了高水平的肿瘤坏死因子-α、IL-1β、IL-6 和NF-κB 的激活,I-κBα 水平显著降低,这些结果提示MSU 晶体刺激诱导Mϕ 向M1 型极化,随后促进促炎介质的产生[17,22]。
2.JAK/STAT 信号通路:Janus 激酶(Janus kinase, JAK)/酪氨酸激酶受体和信号转导和转录激活因子(signal transducer and activator of transcription,STAT)信号通路不仅与Mϕ 的炎症相关,也参与Mϕ极化过程。 IFN-γ 介导的JAK/STAT 信号通路促进M1 型表达NOS2,增加IL-1、IL-12 等促炎性细胞因子的释放[23]。 在痛风性关节炎中,MSU 晶体可诱导小鼠滑膜组织中IL-6 分泌增加,促使p-JAK2 和p-STAT3 的表达水平明显升高,可引起Mϕ 向M1型极化,并且早期可诱导部分Mϕ 向M2 型极化,提示JAK2-STAT3 信号参与了MSU 晶体诱导的早期炎症过程中Mϕ 向M1 型和M2 型的极化过程[17]。STAT6 是Mϕ 极化到M2 型的最重要的转录因子之一,磷酸化的STAT6 进入细胞核,介导了Mϕ 的脂质代谢,促进M2 相关基因的转录,诱导Mϕ 向M2 型极化[13]。
3.NLRP3 炎症体信号通路:炎性小体是一种大的胞质多蛋白复合体,控制促炎性细胞因子的成熟和分泌,包括IL-1β、IL-18 和细胞凋亡。 NLRP3(NOD-like receptor family pyrin domain-containing protein 3)炎症体是一个多蛋白复合体,包括NLRP3、凋亡相关斑点样蛋白(apoptosis-related speckle-like protein,ASC;包含CARD caspase 激活和招募结构域) 和半胱氨酰天冬氨酸特异性蛋白酶-1(caspase-1)[24]。 NLRP3 识别MSU 晶体信号后发生活化,暴露出核苷酸结合寡聚化结构域(NACHT),进一步募集ASC 和caspase-1 形成炎性小体,切割前体IL-1β,使其成熟后分泌至胞外引发炎症级联反应[25]。 在MSU 晶体诱导的GA 大鼠模型及滑膜细胞中均发现NLRP3 表达上调并且MSU 晶体诱导的GA大鼠模型中NLRP3 炎性小体的高表达促进了Mϕ 向M1 型极化[26,27]。
五、巨噬细胞极化与痛风性关节炎治疗
NF-κB 信号通路是促进Mϕ 向M1 型极化的主要通路之一,抑制其活化可以阻碍Mϕ 向M1 型极化,以缓解GA 的炎症。 四妙丸是中医临床上经验性用于治疗GA 的经典中药方剂且具有体内外抗炎活性,即可通过失活NF-κB 信号通路抑制Mϕ 向M1型极化来抑制GA 的炎症发展,又可以通过诱导M2型极化来促进炎症的消退[17]。 在痛风性关节炎急性期,剧烈的疼痛可以触发神经免疫系统,MSU 晶体可以上调背根神经节中分泌卷曲相关蛋白2(secreted frizzled-related protein 2,sFRP2)分泌,并通过调节NF-κB 信号通路抑制MSU 晶体诱导的炎性细胞浸润和M1 型极化,提示sFRP2 可以作为减轻疼痛和降低不良反应的区域治疗方法的新靶点[22]。
沉默信息调节蛋白1(silencing information regulator protein 1,SIRT1)是依赖于烟酰胺腺嘌呤二核苷酸(nicotinamide adenine dinucleotide,NAD)的第三类组蛋白去乙酰化酶(histone deacetylase,DAC),具有抗炎作用。 白藜芦醇作为Sirt1 的激活剂,可以抑制TNF-α 诱导的成纤维细胞炎症,在痛风性关节炎患者中, Sirt1 通过激活PI3K/Akt/STAT6 通路减轻尿酸盐诱导的炎性反应,激活STAT6 在理论上可能促进M2 型极化,从而发挥抗急性痛风性关节炎的作用[13, 28]。
秋水仙碱作为治疗痛风的经典药物,其主要作用是秋水仙素与α 和β-微管蛋白(tubulin,TUB)结合,形成微管-秋水仙素复合体,防止微管的形成,进而阻碍细胞因子和趋化物质的产生,调节内皮细胞的黏附蛋白表达,抑制IL-1 诱导的L-选择素的表达,调节细胞因子的成熟和释放,并减少中性粒细胞对细胞因子的趋化,秋水仙碱可以阻断MSU 晶体激活NLRP3 炎症体的过程,从而阻止前IL-1β 的加工和IL-1β 的释放,达到抑制Mϕ 向M1 型极化的过程[29]。 阿那白滞素(anakinra)、利纳西普(rilonacept)、康奈单抗(canakinumab)作为IL-1R 拮抗剂,可有效抑制NLRP3 炎性小体通路,抑制Mϕ 向M1 型极化,且对秋水仙碱等传统药物不耐受的患者推荐应用[30]。
六、展 望
在痛风性关节炎中,NF-κB、JAK/STAT、NLRP3炎性小体等多种信号通路参与Mϕ 的极化,不同的药物可以通过影响Mϕ 极化通路而发挥抗炎作用。 尽管Mϕ 极化在痛风性关节炎中已经被证实,但是Mϕ极化过程中,炎症的M1 型和抗炎的M2 型位于极化轴的两端,并且两者之间存在许多具有混合促炎和抗炎特性的Mϕ/M1/M2 型。 当受到环境、代谢、细胞因子分泌等影响都可能会导致Mϕ 从M1 型转变到M2型,反之亦然,或者变成两种细胞的混合体,这也突出了Mϕ 极化的可塑性和复杂性,在痛风性关节炎中仍需开展进一步研究。
猜你喜欢
现代财经-天津财经大学学报(2022年5期)2022-06-01
航天电子对抗(2022年2期)2022-05-24
医学概论(2022年4期)2022-04-24
人工晶体学报(2021年10期)2021-11-26
北京航空航天大学学报(2021年9期)2021-11-02
现代临床医学(2021年3期)2021-07-16
中华养生保健(2020年4期)2020-11-16
航天电子对抗(2019年4期)2019-06-02
中国民族民间医药·下半月(2014年4期)2014-09-26
