
纳米铝热剂的微观模拟研究进展
2022-11-04 02:18:50王亚军冯长根
火炸药学报 2022年5期
崔 巍,王亚军,甘 强,冯长根
(北京理工大学 爆炸科学与技术国家重点实验室,北京 100081)
引 言
铝热剂是一种古老的烟火剂,最早为按照一定比例配置的铝粉与氧化铁粉末的物理混合物[1]。随着科学技术的不断发展,铝热剂的概念、范畴也不断得到扩展和推广,现在通常指点燃时可以放出大量热的燃料(如金属)与氧化剂(如金属氧化物、含氧酸盐、氟聚物等)的混合物或复合物。传统铝热剂多通过将燃料与氧化剂进行物理混合来制备,其组分尺度通常在微米量级,燃烧速率和能量释放速率较低[2],极大地限制了材料性能的发挥以及其在更广泛领域的应用。纳米铝热剂是近年来快速发展起来的一类新型纳米含能材料,又被称为超级铝热剂,是指由粒径为纳米尺度的燃料和氧化剂经复合处理而得到的纳米级复合含能材料[3],由于组分颗粒尺寸大大缩小,使得材料燃烧放热性能大幅提高,相关性能指标最大可提高1000倍以上。其储能放热原理是利用了燃料与氧化剂之间的铝热反应,具有能量密度高、燃烧速率快(数千米/秒)、反应传播临界尺寸小(数微米)等优点[4-5],在微含能器件(微点火[6]、微起爆[7]、微推进[8]等)、高能添加剂[9]等领域展现出良好的应用前景,已成为含能材料领域一个重要研究方向。
目前,纳米铝热剂的实验研究主要集中在微观结构调控与宏观性能表征上,主要有以下几个方面:(1)基本热力学性质及燃烧性能研究,主要采用差示扫描量热法(DSC)、热重分析(TGA)、X射线衍射(XRD)等技术获得材料的放热性能、起始反应温度、热稳定性等[10-12];(2)铝热反应机理研究,主要通过XRD图谱进行分析,检测不同反应时间下生成的产物,以最小自由能原理推测可能的反应历程[13];(3)特殊载荷条件下的燃烧及相关研究,研究条件有高压、冲击、激光等[14-17];(4)材料改性相关研究[18-19]。经过多年的研究,在纳米铝热剂的制备、性能、理论基础和应用等方面均取得了显著的进展[20],但同时也面临着很多棘手的问题,如对纳米铝热剂活性保护、反应及能量释放过程中活性铝的完全氧化等是纳米铝热剂目前面临的最大挑战之一。此外,纳米铝热剂点火、传播机理以及反应传播不同阶段的差异与主导因素等相关研究较少,而实际上,纳米铝热剂的反应过程十分复杂,如涉及反应物相变[21-22]、氧化剂分解[23]、原子级扩散[24]以及在快速和高温反应中难以通过实验测量的其他现象。因此,对纳米铝热剂反应过程继续进行深入研究和理解,便于得到简化或理想的单因素模型,有助于材料设计,对于宏观结构与燃烧性能的把控也有着重要的现实意义。
含能材料的理论计算研究一般可分为静态计算和非平衡态计算两大类,前者可采用周期性密度泛函理论(DFT)来计算模拟含能材料的静态结构,研究晶体性质、缺陷等[25-27],而后者可采用经典分子动力学(MD)、半经验从头算分子动力学(AIMD)方法研究含能材料的热分解,剪切应变,激光、冲击波等引发的反应机制[28-30]等。目前,针对纳米铝热剂的相关分子动力学研究较少,现有研究重点集中在反应物静态界面性质、反应机理、反应行为、反应特性等方面。研究人员根据不同模拟方法的并行计算效能与硬件条件并结合研究目的,一般选择周期性密度泛函理论计算、基于ReaxFF力场的经典分子动力学模拟和半经验从头算分子动力学模拟3种方法。
本文主要对采用微观模拟方法研究纳米铝热剂的静态界面稳定性、反应机理、反应行为和反应特性等工作进行综述,分析各种因素(如加热速率、反应温度、反应物间距等)对反应特性(放热特性、点火延迟)的影响。最后对采用微观模拟方法研究纳米铝热剂的未来发展方向和趋势进行展望。
1 纳米铝热剂的微观模拟方法
含能材料宏观层面的反应特性本质上由其微观结构决定,在微观尺度上对材料进行研究是明确其热力学性质、反应路径的重要途径,因此,根据研究对象和研究目标选择合适的微观研究方法十分重要。微观模拟方法根据是否考虑电子作用可分为分子模拟方法和量子化学计算。分子模拟主要包括基于力场的分子动力学和蒙特卡罗(Monte Carlo,MC)方法;量子化学计算则通过数值求解薛定谔方程来计算原子、电子信息。对于多电子体系,量化计算直接求解薛定谔方程需要很大的代价,一般需合理简化和近似才能得到有用的信息。目前,最简单实用的近似方法为单电子近似,常用密度泛函理论方法和Hartree-Fock近似。近年来,密度泛函理论发展趋于成熟,在材料模拟中占据主流地位,尤其在含有金属原子的体系中更有优势。对于纳米铝热剂反应体系,由于涉及化学反应,而基于概率统计理论的蒙特卡洛模拟无法得到系统内的时空演化路径,无法描述系统的动力学过程,因此不被使用。目前常用的微观模拟方法主要有ReaxFF反应分子动力学模拟、周期性密度泛函理论计算以及从头算分子动力学模拟3种[31-33],见图1。

图1 3种微观模拟方法示意图Fig.1 Schematic diagram of three micro-simulation methods
1.1 密度泛函理论计算
基于密度泛函理论的计算主要集中在静态电子结构和热力学性质等相关研究上。其中电子结构包括能带、态密度和电荷密度等,可以对材料的某些物理及化学性质进行探讨和洞悉;力学性质包括弹性常数、弹性模量以及应变等;基于第一性原理可以精确计算体系基态能量的优势,易于得到体系的热力学参数,还可以计算形成能、表面能和吸附能等。针对铝热反应体系,反应组分间存在界面,其物理化学性质引起了研究者的广泛关注,而周期性密度泛函计算方法可以精确计算体系的表面能、形成能以及各种电荷密度等性质,因此成为研究铝热剂组分界面性质的有力工具[31, 34-37],其中VASP(Vienna Ab-initio Simulation Package)程序包和CASTEP(Cambridge Sequential Total Energy Package)程序包被广泛使用。
1.2 ReaxFF反应分子动力学模拟
分子动力学计算的有效性、合理性很大程度上取决于所选取力场的适用性。在最早的纳米铝热剂分子动力学研究中,Tomer等[38-39]通过自建原子间势函数,利用经典分子动力学方法研究了室温下纳米晶体α-Fe2O3+fcc-Al的力学性质、反应中冲击波速度和反应粒子速度,由于其使用的EAM势(Embedded Atomic Method,嵌入原子法)不能反映原子周围配位环境改变对电子密度的影响,因此未能描述化学反应。通过键级来描述原子间相互作用的ReaxFF反应力场则可以准确反映体系中的化学反应过程[40]。目前,ReaxFF反应力场已经成功应用于高分子材料、含能材料模拟等多个领域[41-43]。2012年,Shin等[42]和Narayanan等[44]分别发展了包含Al、Fe、Ni、Cu等元素和包含Al、Si、Li等元素的ReaxFF力场,使含铝体系的ReaxFF分子动力学研究成为可能。同年,宋文雄等[45]采用ReaxFF反应力场计算了单质铝的多种物理性能,并与嵌入原子法(EAM)计算值、DFT计算值及实验值进行了对比,发现ReaxFF力场对材料含金属键部分的能量变化和原子扩散可以进行合理的描述;应用键级可以较好地描述具有方向性的共价键;固体材料燃烧过程中的传热机制热传导较接近实际情况,可用于描述金属铝反应过程中的能量释放和传播情况。2014年,张金平等[46]第一次采用ReaxFF分子动力学方法模拟了层状结构的Al/SiO2纳米铝热体系,之后陆续出现了基于ReaxFF反应力场的分子动力学模拟研究,如Al/SiO2体系[47]、Al/Fe2O3体系[33, 48]、Al/NiO体系[49-50]和Al/MoO3体系[51]等。基于ReaxFF反应力场的分子动力学模拟的原子数在几千到上万个,模拟时间可达到几个纳秒,能较为完整地分析铝热反应过程,但ReaxFF势参数在准确性和可移植性方面存在一定局限,易受元素种类的限制[28],许多铝热反应体系还不能进行有效模拟。
1.3 从头算分子动力学模拟
一般来说,基于经验力场的分子动力学方法在势函数的精确性与可移植性等方面存在较大局限性,随着计算机硬件性能的提高和计算软件的发展,在密度泛函理论框架下的分子动力学模拟是从头算分子动力学模拟(AIMD)普遍采用的方法之一,在材料设计、合成、模拟等方面取得了明显的进展。从头算分子动力学通过波函数来描述电子的运动状态,可以在不需要任何先验参数情况下准确计算分子、原子的理化性质,包括坐标、受力、能量、电荷等,计算精度较高,常被作为标准数据来使用。从头算分子动力学方法主要基于以下3个假设:(1)忽略系统的核量子效应;(2)系统满足轨道近似(即单电子近似);(3)系统满足绝热近似。在空间尺度极小且精度要求高的场景,如少量分子体系的化学反应路径研究、动力学参数计算,从头算方法表现出了独特的优势。基于以上特点,2008年出现了最早的纳米铝热剂从头算分子动力学模拟[52],并在此研究基础上提出了金属-氧翻转机制[32]。之后,研究者们采用从头算分子动力学方法对纳米铝热剂体系进行了相关研究,建模大都为Al/金属氧化物/Al类三明治结构,原子数目较少,时间尺度在100ps以内,不能较为完整地分析铝热反应过程。
2 反应机理模拟
铝热反应本质上是氧化剂对金属(铝)的氧化,基于实验及数值模拟研究,已经提出了一些反应机理[53-54]。微观模拟方法对反应机理的提出与验证起到了关键的作用,一些对覆盖有氧化层的纳米铝颗粒的分子动力学研究可以很好地描述纳米铝颗粒氧化前期的一些机理,包括扩散-氧化机理、熔融-扩散机理和离子扩散机理等,而基于从头算分子动力学提出的金属-氧翻转机理也经过了验证。
纳米铝颗粒通常以壳-核结构稳定存在,作为纳米铝热剂的重要组分,其有着卓越的性能(如高能量密度,高感度,高反应温度等),引起了研究者广泛的关注。扩散-氧化机理是由Rai和Park等[21, 55-56]提出的。Rai等[56]采用高分辨率透射电镜对铝粉(20~30nm)氧化后的形貌进行了分析,发现当环境温度高于1000K时,铝粉中心区域密度降低,呈现出空心结构;铝粉发生氧化时,核内铝原子向外扩散,氧原子向内扩散。Zeng等[24]通过ReaxFF分子动力学模拟了含氧化层纳米铝颗粒的热诱导反应过程,通过比较不同原子的均方根位移(见图2(a)),证实了覆盖有氧化层的纳米铝颗粒的初始反应主要源于氧原子的向内扩散。Chu等[57]构建了氧化层覆盖的纳米铝颗粒并将其置于氧气环境中进行研究,结果表明,纳米铝颗粒的氧化过程依次经历预热、熔化、铝核氧化和氧化层氧化4个阶段,铝核在核-壳界面熔化,导致核内铝原子向外扩散[见图2(b)]。此外,Chu等[57]和Chakraborty等[58]在有氧化层覆盖的纳米铝颗粒的弛豫过程中均发现了体系势能不断下降的现象,结合模型可视化获得了轻微的氧化扩散图像。Zhao等[59]通过ReaxFF分子动力学模拟研究纳米铝颗粒与炸药(PETN)在加热条件下的相互作用时发现,小粒径的纳米铝颗粒氧化过程以铝离子的扩散为主导,氧化过程分为化学吸附、缓慢氧化和熔融氧化3个阶段,在前两个阶段,O原子向内扩散到Al核心形成一定的氧化层,熔化后的铝原子在最后阶段向外扩散。这些研究均证实了扩散-氧化机理。

图2 (a) 氧化层覆盖的纳米铝颗粒体系内原子的均方根位移随时间变化曲线[24]; (b) ANP四个氧化阶段示意图[57]; (c) PETN/ANP氧化过程纳米铝颗粒的形态演变的剖面图[57]Fig.2 (a) Time evolutions of atomic mean square displacements at the core-shell interfaces[24]; (b) Four oxidation stages of nano-aluminum particles[57]; (c) Profile of the morphological evolution of ANP during oxidation in PETN/ANP[59]
熔融-分散机理描述了带有氧化层壳-核结构纳米铝颗粒的快速氧化过程,由Levitas等[60-61]在2007年首次提出。在极高的加热速率(106~108K/s)下,Al核熔化,其体积膨胀约6%,从而引起核内压力急剧升高,核内外产生巨大压力差(0.1~4GPa),使得氧化层破裂,产生的高拉伸应力使得液相铝分散成小的液相团簇并高速(100~250m/s)飞散,从而引发铝热反应。与扩散-氧化机理不同的是,熔融-分散机理针对的是包裹氧化层的纳米铝颗粒在极高升温速率下的过程,氧化层产生裂纹甚至分散与否,取决于加热速率和壳的厚度,而在加热前期温度低于铝的熔点时,则会发生扩散-氧化过程。目前已有一些关于铝和炸药相互作用的研究[59, 62-64],其中Zhong等[62]采用ReaxFF分子动力学方法研究了从300K加热至3000K过程中纳米铝颗粒在RDX中的裂解机理,有着完整或不完整氧化层的铝颗粒必然会出现裂纹,从微观模拟层面验证了熔融-分散机理,此外研究还表明,氧化壳层越厚,可以承受的由于铝熔化而产生的压力越大。
不同于由压力驱动原子扩散的扩散-氧化机理,基于分子动力学模拟提出的离子扩散机理[65]认为覆盖有氧化层的纳米铝颗粒的点火主要是因为表面的氧化铝层产生了内在电场,加速了Al离子穿过氧化层的扩散速率,并且增强了其扩散强度。这种内在的电场驱动了约90%的Al离子的质量通量。Zhdanov等[66]对纳米铝颗粒的Cabrera-Mott效应进行了理论分析,认为Al离子的扩散不仅由铝核的膨胀引起,而且主要是因为氧化层产生了电场。Zeng等[24]对有氧化层覆盖的纳米铝颗粒在加热条件下的反应进行ReaxFF分子动力学研究发现,在加热阶段前期,氧化层与铝核界面处产生的电场强度最大,而随着温度升高,铝核与氧化层界面处的电场强度逐渐降低,直到2000K时,界面处的电场强度基本为0,此时氧化层完全熔化。
Shimojo等[32, 52]采用从头算分子动力学方法对发生在纳米Al/Fe2O3复合粒子接触界面上的铝热反应进行模拟,提出了金属-氧翻转机制。该机制认为,界面处的氧化还原反应使Al—O键快速形成,随后燃烧波以70m/s的速度及2000K的火焰温度传播,至少持续5ps,氧在反应界面上由Fe2O3体相转移到Al2O3体相中。该机制强化了两相界面处的传质与反应速率,导致了两个阶段的反应过程,在小于1ps时,金属-氧翻转机制使得反应快速进行,之后是原子扩散的缓慢过程。Feng等[67]使用从头算分子动力学对Al/NiO纳米体系的点火和燃烧机理进行研究,通过原子构型的可视化分析了原子在Z轴方向上的位移,也观察到了金属-氧翻转这一现象(见图3)。
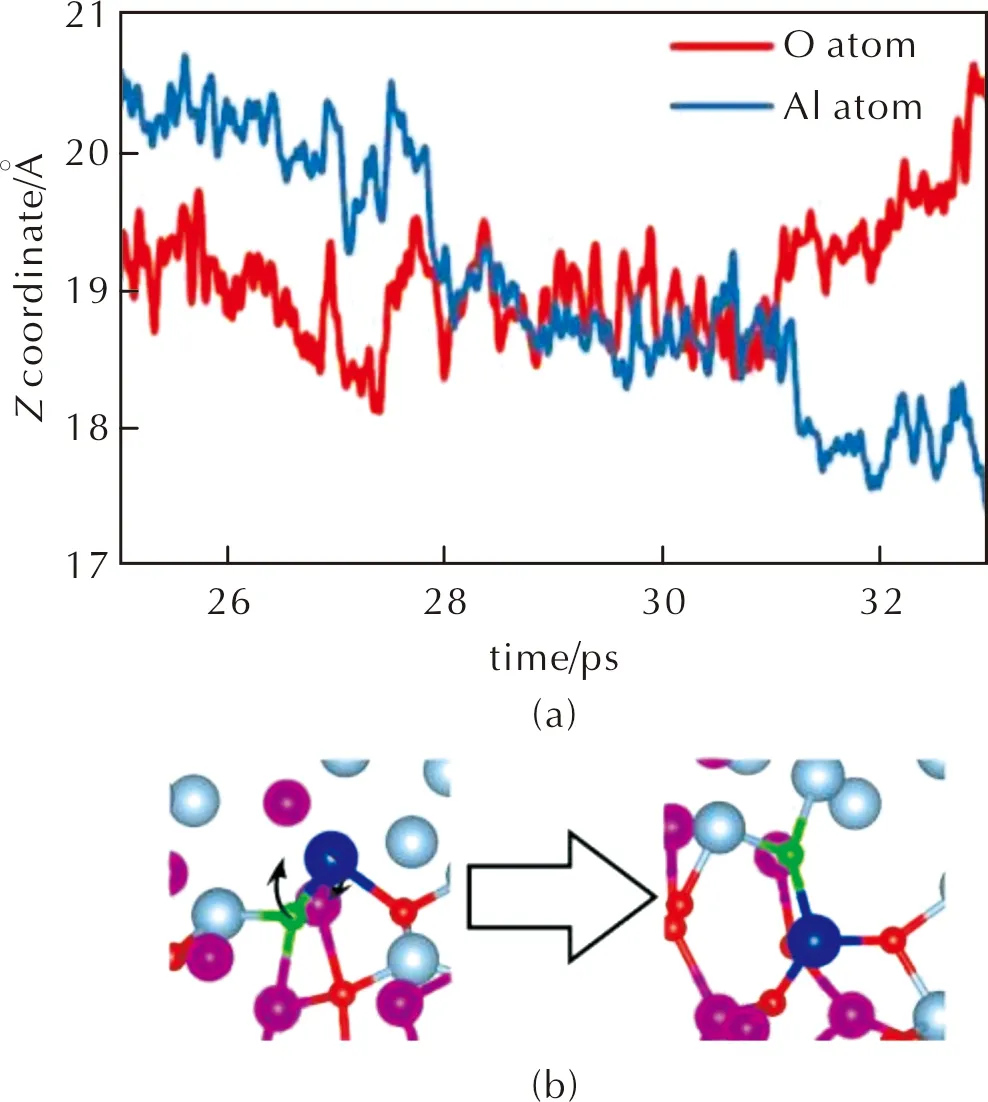
图3 (a) 铝原子与氧原子的Z轴坐标随时间的变化; (b) Al/NiO铝热剂金属-氧翻转机理的原子构型,蓝色和浅蓝色球体表示Al原子,绿色和红色球体表示O原子,紫色球体表示Ni原子Fig.3 (a) Time evolution of the Z coordinate of oxygen and aluminum atoms; (b) Atom configurations of metal-oxygen flip mechanism for the Al/NiO thermite. The light blue and blue spheres are Al atoms, the green and red spheres are O atoms,and the purple spheres are Ni atoms
高表面能赋予了纳米铝颗粒优异的性能,然而也会使其极易相互接触发生粘连团聚,在温度升高时团聚现象极为严重,这也影响了纳米铝热剂的燃烧速度,成为纳米铝热剂目前面临的一大挑战之一。预点火-熔结机理描述的就是纳米铝热剂点火过程中团聚粒子会发生快速熔结的这一现象[53]。Zachariah等分别从反应分子动力学[58]与实验[22]两方面入手研究纳米铝热剂的点火过程,认为纳米铝颗粒前期受离子-扩散机制[65]主导,氧化层使粒子的内部产生电场,驱使Al离子进入氧化层,氧化层在低于其熔点的温度下发生熔化,之后表面张力驱使熔融液态粒子发生融合。在铝热反应发生之前,反应物粒子最初的尺寸和形态就发生了明显的变化,这解释了纳米粒子未能如预期的那样发生快速反应的原因。值得一提的是,包覆是一种改变ANP表面性能的有效措施,包覆层不仅会保护氧化层内的活性铝,提高纳米铝颗粒的抗氧化性能和耐腐蚀性,还可以改善其表面电荷性质与表面化学反应特性,在纳米铝热剂的应用中具有重要意义。Liu等[68-70]对纳米铝颗粒包覆乙醇、乙醚等有机物过程进行了研究,分析了包覆后纳米铝颗粒的常温抗氧化能力、团聚过程与燃烧过程。研究表明,两种完全包覆后的纳米铝颗粒均可以保存至少70%的具有氧化潜力的铝原子,在300K下乙醇包覆层可以主动捕捉外界的氧气分子,同时,体系并未有CO2、H2O等氧化产物生成,这说明乙醇包覆层可以提高纳米铝颗粒在常温下的抗氧化能力;在燃烧模拟中,无有机物包覆的纳米铝颗粒燃烧温度最低为3581K,只能维持颗粒的液相燃烧,而有机物包覆的纳米铝颗粒燃烧温度可超过7000K,有机物释放的热量足以促进体系进入气相燃烧,这表明有机包覆层可以显著提升纳米铝颗粒的抗氧化性能与燃烧能力;对团聚过程的模拟显示,有机包覆层可以有效阻碍颗粒的团聚,粒径为4nm的纳米铝颗粒即使在1000K下也没有观察到团聚现象[70],这是因为碳原子与氧原子占据了纳米铝颗粒表面上低配位的铝原子的结合点位,与未包覆颗粒相比表面势能较低。此外,研究人员发现氟聚物包覆纳米铝颗粒的方法也可以有效解决纳米铝粉易烧结的问题[71],并且显著改善其燃烧性能。在最新的研究中,Liu等[72]分别对有PTFE(聚四氟乙烯)涂层包覆的纳米铝颗粒和无包覆层的纳米铝颗粒在氧气与PTFE环境中的烧结与燃烧过程进行了ReaxFF分子动力学模拟,研究表明PTFE在任何温度下均可以阻止纳米铝颗粒发生烧结,在加热的初期阶段,PTFE与氧化层紧密接触,同时分解为小Al—F团簇,将纳米铝颗粒推开;进入燃烧阶段后,由于PTFE与纳米铝颗粒的相互作用而导致有效铝含量减少;此外,在相同温度下,发现Al—F化合物的扩散程度高于Al—O化合物,这可以阻碍纳米铝颗粒间的接触传质。纳米铝热剂反应机理微观模拟研究情况总结于表1。

表1 纳米铝热剂反应机理微观模拟总结Table 1 Summary of micro simulation of reaction mechanism of nanothermite
相关反应机理虽然可以较好地描述特定条件下的反应规律,但都有各自的局限性,且侧重于解释单一问题,而无法解释纳米铝热剂材料的复杂反应过程。针对覆盖有氧化层的纳米铝颗粒的氧化分子动力学研究,虽然可以对铝氧化反应机理进行较好的描述与验证,但其在建模层面多将纳米铝颗粒置于氧气或炸药环境中,研究对象并非铝热剂,而且炸药的热分解与氧气分子的扩散引发铝的氧化过程与金属氧化物引起的铝的氧化过程并不相同。纳米铝热剂是一个固相复合体系,氧化剂(金属氧化物)的熔沸点都很高,其点火到燃烧的过程中主要发生凝聚相反应[48],因此,在微观模拟层面需要建立新的模型以尽可能描述真实情况下的铝热反应。
3 界面稳定性模拟
纳米铝热剂主要由燃料和氧化剂组成,金属燃料与金属氧化物氧化剂之间的界面控制着材料的反应温度、反应动力学与低温稳定性,深入了解界面结构的性质对纳米复合材料的制备和性能调控都具有重要意义。在纳米铝热剂应用方面,铝/金属氧化物复合多层膜在微机电系统(MEMS)中有着潜在的应用前景。研究发现,在铝-金属氧化物纳米箔界面处,总是发生界面反应而形成厚度为几纳米的薄层[73],影响纳米铝热剂的稳定性和敏感性。但现有的实验研究手段还无法有效表征界面层,因此,从微观模拟层面理解界面结构具有重要的现实意义。
Lanthony等[36]为了研究界面层的生长过程,采用周期性密度泛函理论计算方法分别研究了Al沉积到CuO(1 1 -1)表面和CuO沉积到Al(1 1 1)表面的过程。首先将单颗Al原子置于目标点位以统计系统能量,获得Al原子在不同点位的吸附能垒,从而得到了Al在CuO(1 1 -1)晶面上的垂直扩散反应路径的能量变化,合理推测出Al原子的渗透路径。结果表明,单个Al原子在CuO表面的吸附是高度放热的(Eb=-5.08eV),同时,预测吸附过程中释放的热量足以克服0.6eV的活化势垒,从而使Al进一步渗透到第一层Cu原子中;之后发现了CuO在Al(1 1 1)表面的自发解离;其后,采用相同的方法分别对Cu原子和O原子进行相应研究。结果表明,在低覆盖率下,CuO层和Al层之间存在较弱的相互扩散,在界面处发生混合,而O原子则留在表面。Kwon等[37]在Lanthony等[36]的研究基础上采用相同的方法研究了Al原子在CuO(1 1 -1)表面的沉积和渗透过程,研究结果与Lanthony等[36]基本相同,Al原子在表面吸附高度放热,热量足以克服Al原子向亚表面渗透的能垒,但Al原子在次表面向更深层渗透过程中略有吸热;研究还表明,在初始渗透过程中,Al原子侵占了次表面的一个Cu原子的位点,且与周围的O原子形成四面体AlO4结构,Cu原子则移动到外表面与O原子混合。Wu等[31]采用周期性密度泛函理论计算方法模拟了Al原子在MoO3(0 1 0)表面的吸附与渗透过程,结果显示,Al原子更趋向于吸附在MoO3(0 1 0)表面的次表面位点,次表面吸附比表面吸附更稳定。结构和电荷分析表明,当将Al原子置于MoO3(0 1 0)晶面上时,会形成强Al—O键并削弱Mo—O键。
Xiong等[35]通过周期性密度泛函理论计算研究了金属层和CuO(1 1 1)衬底组成的纳米铝热剂(Al、Mg、Ti和Zr)/CuO(1 1 1)的几何结构、电子结构和稳定性。结构上的弛豫主要表现在金属与CuO衬底表面O原子和Cu原子之间的成键,离子键和金属键在界面运动中起到了重要作用;通过研究不同金属与CuO体系的电子态密度,发现电子的活动均主要发生在界面处。薛闯等[34]采用周期性密度泛函理论方法研究了Fe2O3(1 0 4)和Fe2O3(1 1 0)表面以及Al/Fe2O3界面的性质,其中Fe2O3(1 0 4)表面具有两种不同类型的结构,一种为表层只有O原子暴露,另一种是表层有Fe和O两种原子暴露,结果发现,O原子暴露的Fe2O3(1 0 4)表面比其他Fe2O3(1 0 4)表面更稳定,O原子暴露且次表面为Fe原子的Fe2O3(1 1 0)表面更稳定,原子暴露表面形成的Al(1 1 1)/Fe2O3(1 0 4)界面(AFS1)和Al(1 1 1)/Fe2O3(1 1 0)界面(AFS5)具有较大的界面黏附功,通过比较差分电荷密度验证了这一点。
上述对纳米铝热剂界面结构的研究均采用了密度泛函理论计算方法,通过对吸附能、电子结构等进行研究,分析了界面结构的稳定性。虽然没有化学动力学过程,但仍然可以为纳米铝热剂相关分子动力学研究建模提供指导,对纳米铝热剂的材料设计、性能优化以及工程应用提供理论支持。然而目前相关研究还较少,需要进一步扩展研究体系,针对不同的研究目标建立不同的模型,尽可能为分子动力学模拟与实验研究奠定基础。
4 反应行为模拟
纳米铝热剂的化学反应式可以简单描述为:
式中:MaOb为金属M的氧化物。
上述化学式仅笼统地表示了反应物到生成物的过程,而真实铝热反应过程则相当复杂,如反应中存在反应物相变、氧化剂分解、原子级扩散等,以及各种在快速和高温条件下难以通过实验观察到的现象。尽管研究者已经采用分子动力学方法对铝热反应过程进行了一些研究,但对其反应行为的理解还非常有限,以下分别从原子扩散角度和原子间成键角度分析铝热反应过程。
4.1 原子扩散行为
不同于传统的含能材料反应存在分子自身热分解与凝聚态下的热分解,研究表明,铝热反应过程由原子的扩散程度所主导[47],因此,明确原子的扩散行为是理解纳米铝热剂反应行为的重要途径。在早期的铝热体系微观研究中,由于对纳米铝热剂的具体点火机制缺乏共识,Shimojo等[32, 52]就纳米Al颗粒破裂后、熔融Al团簇与金属氧化物作用引发铝热反应这一过程(如图4所示),通过从头算分子动力学方法推测Al/Fe2O3界面体系的铝热反应可以分为两个放热阶段:(1)在Al/金属氧化物界面处由金属-氧翻转机制主导的快速反应;(2)原子扩散运动主导的慢反应。2013年,Wen等[74]在研究Al/NiO纳米铝热剂的反应时,为了进一步明确Al/Ni相的形成过程,采用从头算分子动力学方法对Al晶体与NiO组成的纳米线结构进行了模拟,体系从0K升温至1000K后改为绝热条件。研究发现,在5ps后,Al原子通过Al/NiO界面扩散并且与O原子相遇,Al原子的扩散程度大于O原子。同年,唐翠明等[75]在Shimojo等[32, 52]的研究基础上,采用从头算分子动力学方法研究了Al/Fe2O3体系在2000K、NVT系综下的反应过程,统计时间为7ps。研究表明,O原子的均方根位移大于Fe原子和Al原子,由此可知,在铝热反应中O原子的扩散能力大于Fe原子。
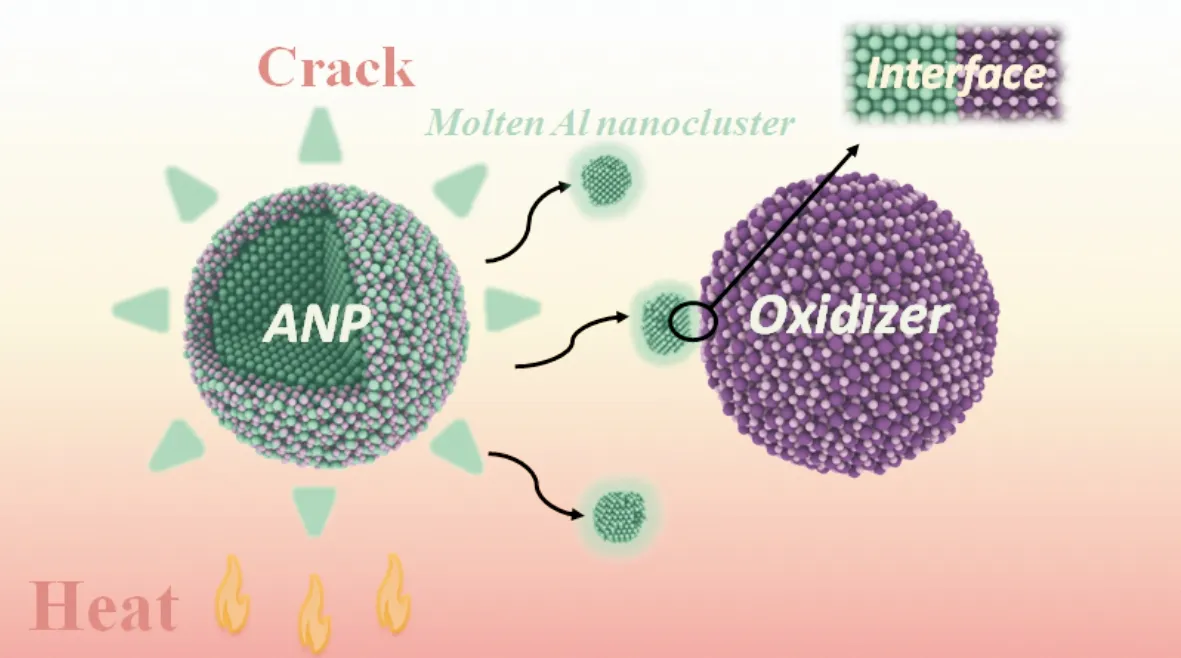
图4 熔融-分散机制主导的纳米铝热剂点火示意图Fig.4 Schematic diagram of the ignition of nanothermite dominated by melt-dispersion mechanism
2014年,张金平等[46]采用ReaxFF分子动力学方法模拟了Al/SiO2体系在不同初始温度下的绝热反应过程,通过分析均方根位移发现,在不同初始温度下原子扩散均经历了两个阶段:(1)原子扩散较为缓慢,属于固体扩散范畴;(2)原子扩散很快,均方根位移随时间线性增加,此时体系已熔化成为液相。之后,Zhang等[47]对相同体系进行了低温诱导铝热反应研究,通过分析扩散系数发现,初始温度越高,体系内原子的扩散系数越大,并推测铝热反应过程由原子的扩散程度所控制。Zeng等[49]采用ReaxFF分子动力学方法研究了蜂窝状Al/NiO体系在加热条件下的反应过程,通过分析均方根位移发现,反应前期体系内原子的均方根位移趋于平缓,之后线性升高,这与张金平等[46]研究结果相似,此外,还发现Al原子的扩散程度大于NiO。
Tang等[76]对Al/CuO纳米界面在不同温度下的反应行为进行研究,采用CASTEP程序包进行了NVT系综下的从头算分子动力学模拟,发现界面处近Al层的CuO中的Cu原子的均方根位移最大,最易与Al原子结合形成Al-Cu合金,推测是因为Al-Cu合金的熔化温度低于纯Al的熔化温度,Al-Cu合金在界面处容易发生反应。Xiong等[77]采用从头算分子动力学方法研究了Al/CuO体系在不同初始温度(830K、1000K、2000K和3000K)下的反应过程,结果表明,温度越高,原子变化越快,特别是当温度高于2000K时,Cu原子开始渗入Al金属层中,Cu层最终完全被3000K的Al原子掺杂。Xiong等[78]在后续工作中,根据原子轨迹可视化分析推测了反应前期O原子的扩散过程(见图5),认为氧化还原反应释放的能量以及Cu层形成空位维持了Al原子的连续扩散运动过程。
图5 从头算分子动力学模拟中不同时刻Al/CuO纳米铝热体系的反应过程Fig.5 Interfacial reaction process of Al/CuO nanolaminate observed at different time during the AIMD simulations
Feng等[67]在Shimojo等[32, 52]的研究基础上,在NVE系综下对Al/NiO纳米界面体系进行从头算分子动力学模拟,从微观角度证实了Al/NiO纳米铝热剂的二次放热特性。Al/NiO铝热剂的反应可以分为5个阶段,即初始阶段、界面反应阶段、NiO晶格破坏阶段、体相反应阶段和反应完成阶段,其中两次放热对应界面反应阶段和原子扩散主导的体相反应阶段,两个放热阶段与Shimojo等[32, 52]研究结果相似,在界面反应中,金属-氧翻转机制降低了铝热反应中氧化层对体相反应阶段传质的不利影响。
4.2 原子间成键行为
研究含能材料原子间的成键行为对明确化学反应中间产物与基元反应有着重要的意义。基于ReaxFF反应力场的分子动力学方法和从头算分子动力学方法在分析反应中化学键的生成与断裂方面有很大的优势。对于炸药材料,可以根据ReaxFF力场的经验键级截断半径推测炸药分解过程中出现的中间产物和基元反应[79],而纳米铝热剂主要涉及凝聚相反应,产物多为凝聚相且成分复杂,气态产物占比很小,因此目前还无法分析中间产物和基元反应。然而,研究者可以通过分子动力学模拟分析键重叠布居、径向分布函数等,统计反应过程中的化学键数量,从而获得有用的化学键信息。
Shimojo等[52]采用从头算分子动力学在2000K的NVT系综下研究了熔融Al/Fe2O3体系界面处发生的初始氧化反应,分析了界面处所有氧原子、Fe—O键、Al—O键的键重叠布居,见图6(a)。在0.2ps前,氧原子位于Fe2O3侧,0.2ps之后逐渐向金属Al侧迁移,随着Al—O键重叠布居的增加,在0.45ps之后Fe—O键重叠布居变为0,预示界面处O原子只与Al原子成键,界面处Al原子的移动由于化学键的变化而触发。在之后的研究中,Shimojo等[32]基于键重叠布居的研究提出了金属-氧翻转机理。唐翠明等[75]在采用从头算分子动力学模拟研究纳米Al/Fe2O3体系时,通过统计0~7ps内化学键的变化讨论了原子的扩散程度,见图6(b)。随着时间的增加,Fe—O键数量减少,Al—O键和Fe—Fe键的数量增加,表明Al/Fe2O3纳米体系铝热反应中发生了氧化还原反应。在张金平等[46]对Al/SiO2体系的ReaxFF分子动力学研究中,通过分析径向分布函数发现,铝热反应前期Si—O键占主导地位,随着时间的增加,Al和SiO2内部的稳定性逐渐破坏,生成了稳定的单质Si和铝氧化物。
在相关实验研究中,铝热剂是否发生分解释放出O2引起了研究者的关注。Zhou等[23]采用温度阶跃飞行时间质谱仪表征了Al/(Fe2O3、CuO、ZnO)纳米复合材料的反应过程,分析表明,金属氧化物颗粒在铝热剂中有储氧的作用,不同于CuO作为氧化剂时释放出大量的氧气而导致的高反应活性,Fe2O3释放出少量的O2。Zhu等[48]采用ReaxFF分子动力学研究了Al/Fe2O3体系的铝热反应,通过统计反应中化学键数的变化描述了氧化还原反应过程,根据O—O键数量随时间的变化程度判断不同反应物间距下氧化剂是否分解产生O2。结果表明,反应物间距为2nm时,1900K下体系释放出14个O2分子,此时体系趋向于多相反应,当反应物间距为1.5nm、1450K时,体系释放出2个O2分子,反应趋向于液相反应,当反应物间距为1nm、600K时,没有O2分子释放,此时反应趋向于固相反应。

图6 (a) (上部)与氧原子相关的全部和部分键重叠布居。黑色、红色和蓝色曲线分别代表(下部)体系在不同时间的原子构型,绿色、红色和灰色球体分别代表Fe原子、O原子和Al原子[52]; (b) Fe—O键、Al—O键和Fe—Fe键数量随时间的变化[75]; (c) 三角形Fe2Al合金结构和四面体Fe3Al合金结构[33]Fig.6 (a) (Up) Time evolution of the total and partial SBOP,associated with an oxygen atom. The black, red, and blue curves show Oi(t), and respectively;(Bottom) Atomic configurations near the oxygen atom of interest (pointed by yellow arrows) at different times. The green, red, and gray spheres are Fe, O, and Al atoms,respectively[52]; (b) Variation of amount of Fe—O bonds,Al—O bonds and Fe—Fe bonds with time[75]; (c) The triangular structure for Fe2Al clusters and the tetrahedral structure for Fe3Al clusters[33]
之后,Zeng等[49]和Lin等[33]分别对Al/NiO体系和Al/Fe2O3体系反应中的原子间键长分布进行了分析。Zeng等[49]发现,不同的点火温度与化学计量比对化学反应过程有很大的影响,但对原子间键长(Al—Ni键、Al—O键)几乎没有影响。Lin等[33]通过对模拟盒子进行二维切片分析了Al—Fe键长,结果表明,产物中存在大量键长为2.60~2.80Å的三角形结构合金(图6(c)),同时存在一些键长为2.30~2.59Å的四面体结构,说明铁铝金属间化合物主要以Fe2Al团簇形式存在,有少量Fe3Al团簇。
5 反应特性模拟
纳米铝热剂的反应通常可以分为预反应和火焰传播两个阶段[54]。在预反应阶段,外界给予能量激励,如撞击、加热等,此时发生固-固相反应,一旦能量积累到点火阈值,就触发点火,反应进入第二阶段,即自持火焰传播阶段。目前,微观尺度模拟的空间尺度、时间尺度分别可以达到几十纳米和几个纳秒,虽然还不能完整地描述铝热剂反应的火焰传播过程,但是一些ReaxFF分子动力学模拟采用了NVT系综升温加热后改变为NVE系综的控温方式,可以很好地模拟纳米铝热剂加热诱导反应的点火过程,其中NVT系综下的升温对应了外界给予能量刺激的过程,NVE系综下体系内总能量固定不变,发生化学反应时,体系势能下降,动能与温度升高,点火延迟期表现为初始反应温度到体系温度发生突跃时刻的时间,点火延迟期对材料体系感度有重要意义。研究者通过改变反应条件,如反应温度、升温速率、反应物间距等,可以研究不同条件下的反应特性(如绝热反应温度、点火延迟时间、有效反应时间等)。
张金平等[46]对Al/SiO2体系进行ReaxFF分子动力学研究时,首先在NVT系综下将体系加热到初始温度(600、700、800、900、1000和1100K),后改为NVE系综,体系在绝热条件下发生自持放热反应,动能的增大表现为温度的升高,最后达到绝热温度。当初始温度为900K和1000K时,体系温度超过绝热温度且不断升高,通过对NPT系综下Al块体熔化过程进行模拟发现,温度—势能曲线与温度—体积曲线都发生了明显的跳跃,覆盖温度范围为900~1020K,此温度范围内的结构被称为“类液”结构,因此推测,在初始温度为900K和1000K时,铝热反应温度曲线不同于其他初始温度是由于出现了“类液”结构,在此结构中,Al表面的最外一层原子没有晶格的约束,高度无序,原子具有一定的流动性,其他原子保持在完美晶格点附近,在平衡位置做热振动,“类液”结构的自扩散发生在原表面最外层而非垂直方向上,一部分发生在原表面最外层内,而更多的扩散发生在原表面最外层之外[80]。特殊的“类液”结构使Al表面流动的原子与SiO2表面发生铝热反应。体系温度升高,Al层和SiO2内部原子吸收热量,使得Al和SiO2内部由固态转变为液态,分布均匀的Al熔体和SiO2熔体紧密接触,促进了Al原子与SiO2发生强铝热放热反应,导致Al与SiO2反应在1ns时未能达到平衡状态。研究还发现[46],初始反应温度越高,系统达到的绝热温度越高,有效反应时间越短(见表2),且模型在没有氧化层存在的情况下[47]反应温度可以低至300K,见图7(a)。
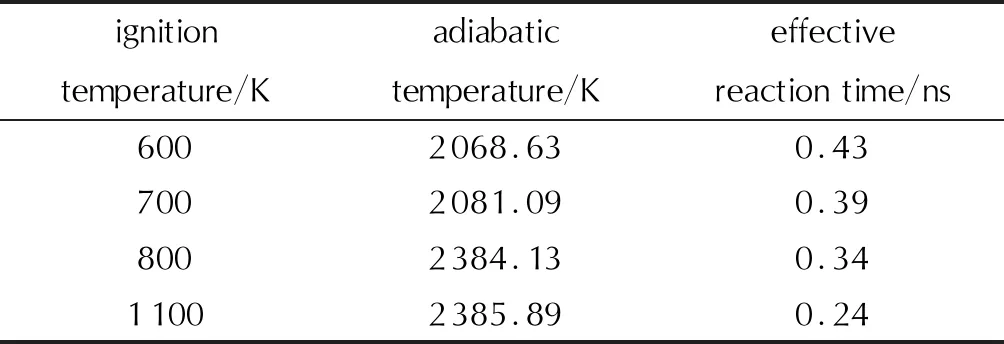
表2 不同初始温度下铝热反应的绝热温度和有效反应时间Table 2 Adiabatic temperature and effective reaction time of aluminothermic reaction at different initial temperatures
Zhu等[48]将不同反应物间距的Al/Fe2O3体系升温至目标初始反应温度,发现反应物间距越大,点火延迟期越长。Lin等[33]分析了不同升温速率、初始温度和反应物团簇间隔下Al/Fe2O3纳米体系的反应特性,在相同加热速率下,初始反应温度越高,绝热反应温度越高,有效反应时间越短;在相同起始反应温度下,加热速率越大,点火延迟期越短,有效反应时间越短。此外,通过点火延迟时间拟合得到了不同升温速率下的活化能(见表3),发现加热速率和初始反应温度越高,引发反应所需的活化能越小,点火延迟期越短。
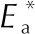
表3 不同点火温度下与加热速率下的点火延迟期Di与活化能EaTable 3 Ignition delay Di and activation energy for ignition at different ignition temperatures and heating rates
Feng等[67]没有采用NVT→NVE的控温方法,而是在NVT系综下将体系温度从10K升温至1500K,通过对Al/NiO铝热中体系势能变化分析其放热特性,其中Ⅱ与Ⅳ阶段对应界面反应和体相反应两个放热阶段,体系势能明显下降,此外还比较了16O与18O同位素对能量释放的影响,见图7(b),其中18O有着较重的质量以及较慢的扩散速度,导致了点火能垒的提高与铝热反应过程整体后移(见表4),这表明O的扩散能力越弱,会导致点火温度越高,点火延迟期越长。
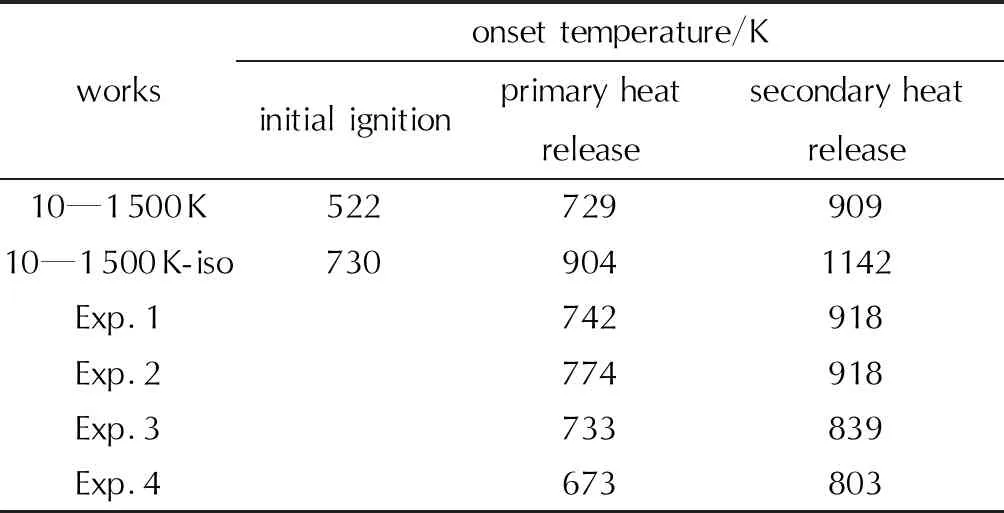
表4 Al/NiO铝热剂在不同反应阶段的起始反应温度[67]Table 4 Onset reaction temperatures of the Al/NiO thermite in different processes[67]
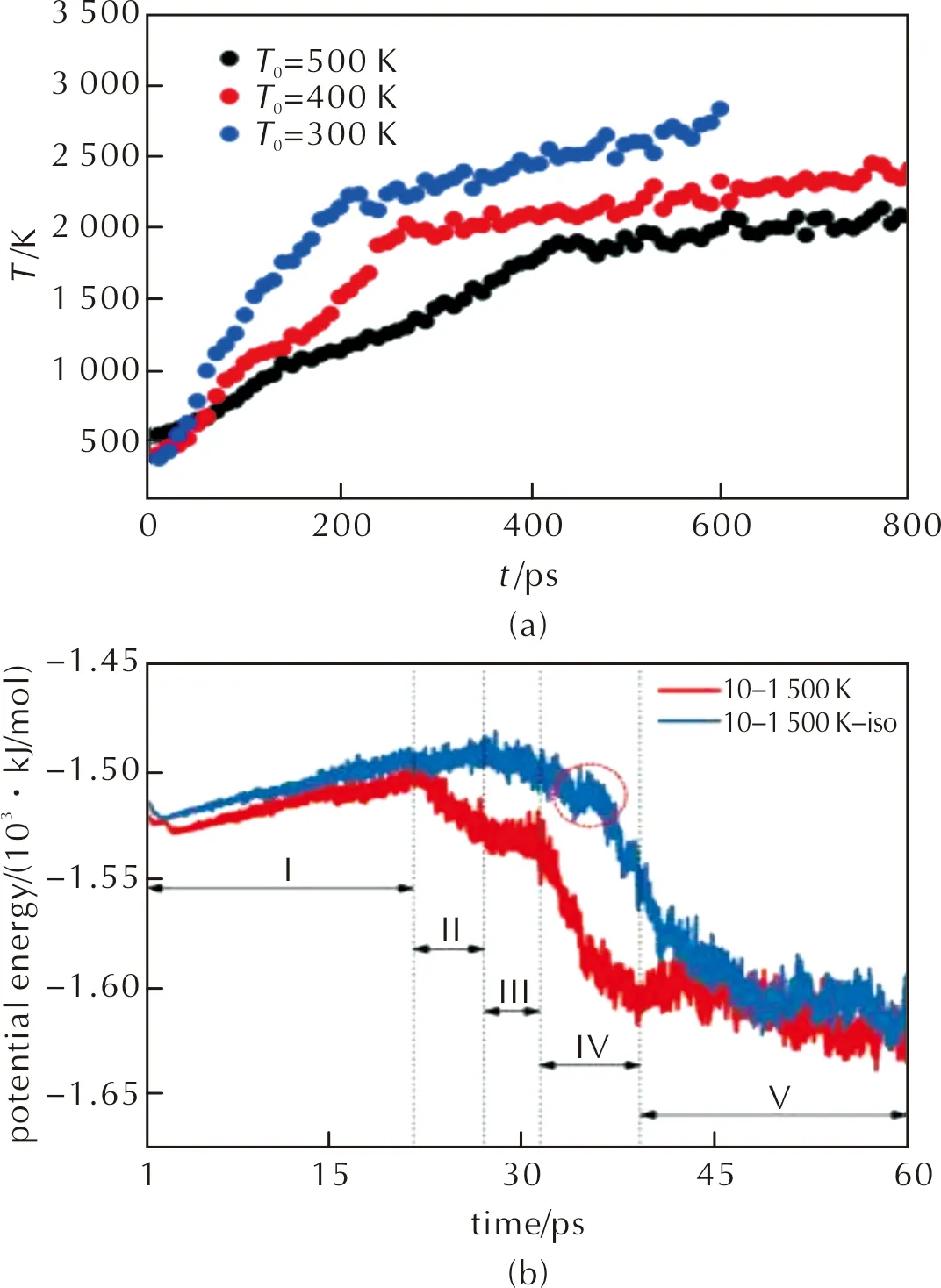
图7 (a) 不同起始温度下Al/SiO2铝热反应过程中的温度变化[47]; (b) 16O与18O的Al/NiO铝热剂在10~1500K模拟中的势能变化对比[67]Fig.7 (a) Temporal evolution of temperature during the thermite reaction for Al/SiO2 with different initial temperatures[47];(b) Comparison of potential energy curves for the Al/NiO thermite in the 10—1500K and 10—1500K-iso simulations[67]
表5总结了目前为止纳米铝热剂微观模拟研究的情况。可以看出,现有纳米铝热剂的微观模拟研究体系较为简单,体系种类偏少,基本局限在Al-金属氧化物界面的相关性质研究;此外,在研究反应行为与反应特性时也只考虑了加热条件,仅文献[81]报道了Al/Fe2O3体系在挤压条件下的模拟点火过程,而其目的是为了研究铝工件在机加工中与铁板发生的冷焊与粘连现象,且研究较早,也不够深入。

表5 纳米铝热剂体系微观模拟总结Table 5 Summary of microscopic simulation of some nanothermite systems
6 总结与展望
目前,微观模拟方法逐渐在纳米铝热剂反应机理、界面结构稳定性、反应行为与反应特性的相关研究中发挥着越来越重要的作用,但是纳米铝热剂的微观模拟研究还较少,尚处于起步阶段,这表现在现有模拟铝热体系种类较少(目前多为金属Al/金属氧化物体系),且各研究仅针对某些特定条件下的反应情况;另外,对不同载荷下的点火机制与燃烧传播过程理论上的理解不够深入与广泛,因此对今后研究做出如下建议和展望:
(1)发挥密度泛函理论计算与从头算分子动力学方法的优势,对金属Al与氟聚物或含氧酸盐所构成的铝热体系进行模拟,深入研究反应机制,同时可以得到针对不同铝热体系的训练集,之后通过拟合对应体系的ReaxFF反应力场,可以基于ReaxFF反应力场研究更大时间与空间尺度的铝热体系,充分发挥不同微观模拟方法的计算优势;
(2)现有纳米铝热剂相关反应机理与微观模拟研究仅考虑升温条件,而在真实使用场景中,铝热反应可在不同种类的载荷下引发,如机械作用、冲击波作用、激光作用等,在今后的研究中,可以探索纳米铝热剂在不同载荷形式下的点火、反应过程,研究其点火、传播机制;
(3)铝热反应研究是一个从微观到宏观、从时间到空间等多个尺度相互耦合与关联的系统性、科学性问题,仅从单一尺度做出的预测与评估都是有局限性的,为了弥补不同微观模拟方法的缺陷,需要发展更为高效的计算方法,用于模拟更多原子数量和更长时间的铝热体系反应过程,如微观—介观—宏观多尺度模拟相结合的方法,开展多尺度下的模拟研究,实现各物理量在不同尺度下的传递与过渡,实现边界条件与计算结果在不同尺度下的耦合。
猜你喜欢
空气动力学学报(2022年4期)2022-08-23 06:51:26
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 06:42:32
当代陕西(2020年13期)2020-08-24 08:22:02
制造技术与机床(2017年5期)2018-01-19 02:49:17
潍坊学院学报(2016年2期)2016-12-01 13:00:11
新闻传播(2015年11期)2015-07-18 11:15:04
浙江大学学报(工学版)(2015年2期)2015-05-30 07:04:53
火炸药学报(2014年1期)2014-03-20 13:17:22
