
PD-1单抗联合R-CHOP方案治疗原发纵隔大B细胞淋巴瘤复合经典霍奇金淋巴瘤1例*
2022-11-03 09:43:02金国凤陶千山沈元元董毅
临床输血与检验 2022年5期
金国凤 陶千山 沈元元 董毅
复合性淋巴瘤(composite lymphoma,CL)是指两种及以上形态学或免疫表型不同的淋巴瘤同时发生于同一解剖部位或瘤块内[1-3]。CL非常少见,而原发纵隔大B细胞淋巴瘤(primary mediastinal large B cell lymphoma,PMBL)复合经典霍奇金淋巴瘤(classic Hodgkin's lymphoma,cHL)的发生率更低,是一种罕见的CL类型,目前相关报道及研究较少[4]。本文报道发生于纵隔的PMBL和cHL组合淋巴瘤病例1例,通过对患者临床特点、病理类型、免疫组化及诊疗经过进行分析,并对相关文献进行复习,提高对这种类型淋巴瘤的认识,并为临床治疗提供新的思路。
资料与方法
1一般资料 患者,男,23岁,2021年5月因“胸痛1月”就诊,完善胸部CT示前纵隔见团块软组织密度灶,周围见肿大淋巴结,增强CT见低密度坏死区,考虑恶性变(见图1),后于全麻下行“正中开胸纵隔肿瘤切除术”。病理检查:灰褐色结节样组织,包膜完整,切面灰白、灰黄色,质软,局部呈鱼肉样,局部呈多房囊性。镜检见胸腺囊肿形成,部分囊壁坏死,纤维组织增生伴大量中性粒细胞、淋巴细胞、组织细胞及嗜酸细胞浸润,且有较多体积大、核型不规则、核深染的异型细胞散在浸润,局部可见中等至大的淋巴样细胞弥漫成片分布。免疫组织化学染色呈部分区域为cHL(结节硬化性):CD20-,PAX5(弱+),CD15+,BOB.1-,Oct-2-,CD30+,CD23-,PD-L1+,CD3-,Ki-67(+);部分区域为PMBL:CD20+,MUM1+,BCL6+,PAX5+,BOB.1+,Oct-2+,CD23+,PD-L1+,CD3-,CD10-,Ki-67增值指数70%。原位杂交EBER(-)(见图2)。病理诊断:复合性原发性纵隔大B细胞淋巴瘤合并经典型霍奇金淋巴瘤。患者转入血液科进一步治疗,查骨髓细胞形态学可见异型淋巴细胞;骨髓病理T、B淋巴细胞呈混合性增生,未见异常淋巴细胞骨髓浸润。PET-CT结果显示:(1)“纵隔肿物”切除术后,术区脂肪间隙模糊,胸骨及临近前纵隔区条形18F-FDG代谢增高,考虑为术后改变;前上纵隔淋巴结影18FFDG代谢略增高,淋巴瘤浸润不能除外。(2)左侧锁骨上、下区淋巴结影,18F-FDG代谢增高,考虑为淋巴瘤浸润(见图3A)。结合患者纵隔内包块直径大于7 cm,分期为Ⅱ期伴大包块;患者无发热、盗汗、体重减轻等B症状,分组为A组;再结合患者年龄、分期、LDH、结外病变、ECOG 评分,综合诊断为组合性PMBL合并cHL(Ⅱ期A组;IPI评分1分,低危组)。同时完善甲状腺功能FT3 4.370 μmol/L,FT4 19.132 μmol/L,TSH 2.51 mIU/L、心肌酶谱、心电图、心脏超声及肝肾功能等,均无异常。
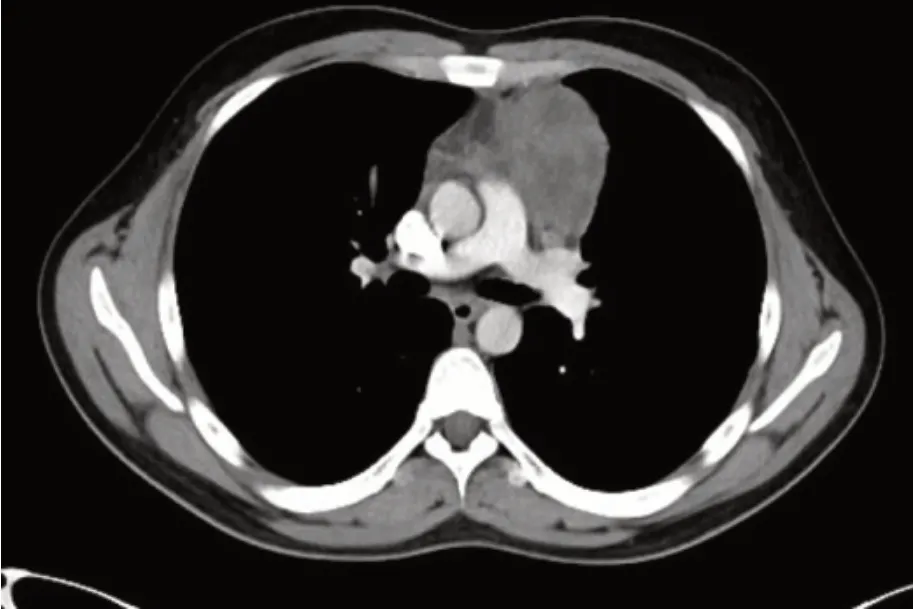
图1 胸部增强CT检查示前纵隔占位
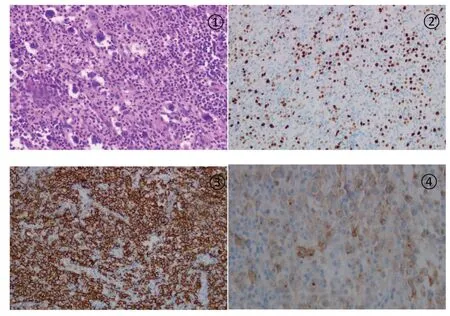
图2 原发性纵隔大B细胞淋巴瘤复合经典型霍奇金淋巴瘤病理
2方法 患者全麻下行“正中开胸纵隔肿瘤切除术”,术后转入血液科综合评估后予PD-1单抗联合R-CHOP(利妥昔单抗+环磷酰胺+阿霉素+长春新碱+泼尼松)方案化疗4周期后完善PET-CT等检查评估,后同方案巩固4疗程。患者治疗通过伦理审核(SLXJS2021-044)并知情同意。
结果
患者PD-1单抗联合R-CHOP治疗4疗程后,PETCT检查评估提示前纵隔病灶及放射性摄取未见异常,8疗程治疗后定期复查随访至今仍处于缓解状态(见图3B)。同时对PD-1单抗导致免疫相关的不良事件进行检测。查心电图及心肌酶等未见异常,FT3 5.112 μmol/L,FT4 17.651 μmol/L,TSH 1.33 mIU/L,没有发生皮肤毛细血管增生症,患者耐受性良好。
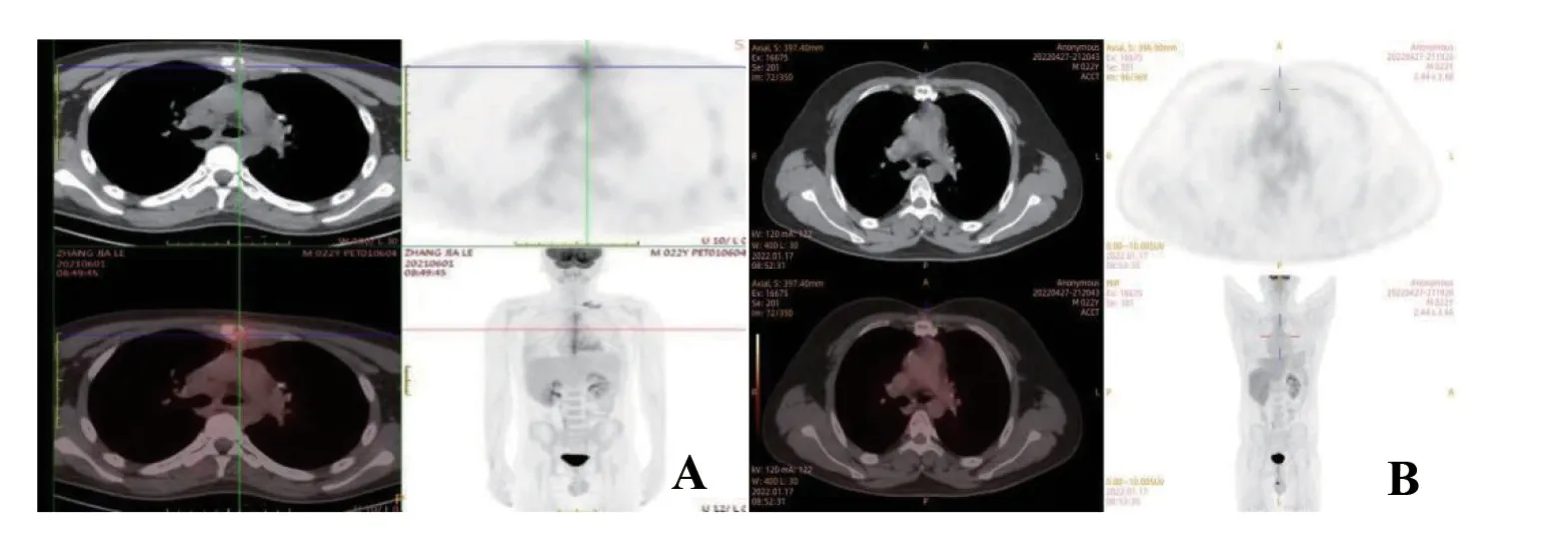
图3 患者PET-CT检查
讨论
患者发生多种组织学类型的淋巴瘤时,称为序贯性或复合性淋巴瘤,不同日期发生不同的淋巴瘤亚型组合为序贯性。最常见的CL是经典HL与低级别B细胞淋巴瘤的组合,序贯性淋巴瘤的主要组合是边缘区淋巴瘤后的弥漫性大B细胞淋巴瘤[5]。但某些惰性淋巴瘤如滤泡性淋巴瘤、慢性淋巴细胞白血病等发展成为侵袭性淋巴瘤,有时在同一解剖部位同时存在则不能称为CL,而是淋巴瘤的转化。CL组织学可分为两大类:(1)两种不同类型的非霍奇金淋巴瘤的组合,(2)霍奇金淋巴瘤与非霍奇金淋巴瘤的组合。大多数CL病例涉及两种不同类型的B细胞淋巴瘤,通常是低级别B细胞淋巴瘤,除与弥漫性大B细胞淋巴瘤相关的滤泡性淋巴瘤外,最常见的CL是与经典HL相关的低级别滤泡淋巴瘤,其次是与套细胞淋巴瘤相关的滤泡淋巴瘤[6-11]。
由于诊断CL技术要求高,其真实发病率可能被低估。通常情况下,更具侵袭性的成分占主导地位,并掩盖了另一种成分,而且由于其定义不一致,不同研究中报道的CL发病率也不同[12-13]。一项在1 000多例NHL病例中进行的研究发现,CL的发生率为1%~4.7%,但取决于临床医生的经验和他们应用的分型系统[8]。诊断目前主要依靠组织病理及免疫组化,而CL可能会增加诊断难度。本例报道的是同时具有cHL及PMBL的病理组织学和免疫表型特征的纵隔CL,组织镜下可见同一区域结构内具有两种不同细胞形态,加上免疫组化标记,形态学及免疫表型上符合CL诊断。另外CL需同灰区淋巴瘤进行鉴别,其在形态学及免疫组化特征介于弥漫大B细胞淋巴瘤与经典型霍奇金淋巴瘤之间[14]。该病例首次病理诊断为特征介于弥漫大B细胞淋巴瘤和经典霍奇金淋巴瘤之间,不能分型,此时与灰区淋巴瘤鉴别显得尤为重要。两者临床特征没有显著差别,但灰区淋巴瘤是部分组织学和免疫组化符合cHL和PMBL,各部分特征所占比例不固定,组织学特点和免疫表型之间没有直接的对应关系,不能确切诊断cHL和PMBL,而CL是同一解剖部位可以明确诊断的两种淋巴瘤。
CL的治疗不同于普通淋巴瘤,迄今为止,CL尚无标准有效的治疗方案。需要针对两种不同的淋巴瘤病理类型制定不同的治疗方案,一致的观点是,占主导地位淋巴瘤应在需要时进行治疗[13],以针对侵袭性较强的淋巴瘤的治疗方案为主,或者对CL病变的序贯治疗[15-16]。本文报道的PMBL和经典HL之间的复合性和序贯性淋巴瘤的资料很少,CL患者的五年生存率为74.8%,与单纯PMBL相当,但是低于单纯cHL[8-17]。PMBCL是一种临床病理特殊类型,与其他淋巴瘤具有相同的临床症状,但缺乏病理学特征,五年无进展生存率和总生存率分别为67%和81%[17]。综合该患者的临床及病理资料,需要针对PMBL进行治疗,PMBL目前主要以联合化疗、放疗治疗为主,部分患者序贯自体造血干细胞移植[18]。虽然DA-REPOCH的完全缓解率高于R-CHOP,但这些患者更有可能出现与治疗相关的毒性反应,2年的生存没有差异[19]。有报道患者接受了6个周期的R-CHOP治疗,以及2个周期的利妥昔单抗维持治疗,并获得完全缓解[8]。PD-1单抗是一种重要免疫抑制分子,基因表达谱研究表明,PMBL和HL有三分之一的共享基因,相关研究已确定这两种疾病有共同驱动突变,并且与PD-L1和PD-L2过表达有关,促进免疫逃避[20-22]。目前PD-1单抗多用于复发/难治的经典HL及PMBL挽救治疗,对于是否同样有效于更前线的淋巴瘤治疗,尚缺少相关研究[20-23]。本例患者HL和PMBL均表达PDL1,确诊后给予PD-1单抗联合R-CHOP方案化疗,4疗程后PET-CT评估达到CR,该患者可以不用进行后续的放疗,如果没有达到CR状态,尚需要放疗巩固。因此,PD-1抑制剂联合R-CHOP对于PMBL复合cHL复合性淋巴瘤治疗是有效的,为临床复合性淋巴瘤治疗提供一个新的思路,后续是否需要进一步行自体造血干细胞移植,仍需多样本的研究。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
传染病信息(2022年3期)2022-07-15 08:24:12
肝博士(2021年1期)2021-03-29 02:32:08
家庭医学(下半月)(2019年11期)2020-01-16 08:39:08
成都中医药大学学报(教育科学版)(2016年1期)2016-01-22 07:24:50
山东医药(2015年14期)2016-01-12 00:39:51
磁共振成像(2015年5期)2015-12-23 08:52:50
分子影像学杂志(2015年3期)2015-12-04 03:29:02
实用肝脏病杂志(2015年5期)2015-12-03 06:28:04
天津医科大学学报(2015年3期)2015-06-05 12:21:49
成都中医药大学学报(教育科学版)(2014年1期)2014-01-19 13:58:28
