
mRNA疫苗质量控制进展
2022-11-01 01:53:24张辉刘建阳毛群颖梁争论徐苗
药学进展 2022年10期
张辉,刘建阳,毛群颖,梁争论,徐苗
(中国食品药品检定研究院,北京 102629)
信使RNA(messenger RNA,mRNA)疫苗是将编码外源目的基因序列通过转录、合成等工艺制备的mRNA通过特定递送系统导入机体细胞,利用机体细胞蛋白质合成机制表达目的蛋白、刺激机体产生特异性免疫反应,获得免疫保护的一种核酸制剂[1]。1961年,Brenner等[2]首次发现mRNA是一种中间遗传物质。1989年,Malone等[3]提出基于mRNA的药物相关概念。1990年,Wolff等[4]将体外转录合成的mRNA直接注射入小鼠骨骼肌细胞中,首次在动物体内成功表达外源mRNA。2005年,Karikó等[5]发现mRNA修饰可降低机体对外源mRNA的免疫反应。近年来,在mRNA修饰[6]、mRNA疫苗递送系统[7-8]等方面研究取得了关键性进展,显著地提升了mRNA翻译效率和稳定性、提高了mRNA疫苗的免疫原性和安全性[9]。和传统疫苗相比,mRNA疫苗具有以下优势:1)mRNA及其递送系统可通过正常细胞代谢实现降解,不会整合到机体细胞基因组中[10]。生产过程中不涉及活病毒的操作,生物安全风险低。2)各种修饰和递送系统可促使mRNA更稳定和实现更高效翻译[7]。mRNA具有自佐剂的效应,具有较强的免疫原性[11-12]。3)完成序列设计和验证后,可直接体外转录产生,具备快速扩大生产至数十亿剂疫苗的能力,可满足重大新发突发传染病暴发时疫苗接种需要[11,13-14]。2021年8月23日,mRNA新冠病毒疫苗BNT162b2(Comirnaty®)获美国食品和药品管理局(U.S. Food and Drug Administration,FDA)批准上市,成为全球首个被正式获批上市的新冠病毒疫苗[15]。目前,全球共34款mRNA新冠病毒疫苗进入临床试验,其中我国7款,数量位居全球前列[16]。
全球对mRNA新冠病毒疫苗大量的需求也对疫苗质控提出了严峻挑战[17]。mRNA疫苗的生产涉及多个生物过程和原材料的加工处理,如大量体外转录(in vitrotranscribed,IVT)、mRNA加工修饰、mRNA脂质体包裹等,生产过程中面临着污染和引入杂质的风险,需研究建立多个和疫苗特性相关的如鉴别、含量、完整性和包封率等全新关键质控参数及其检测方法和质量标准,而残留模板DNA、不完全mRNA、脂质组分降解等检测方法难度较大[18-19]。此外,已上市疫苗需低温保存,疫苗稳定性也为疫苗重要风险之一[20]。我国mRNA疫苗发展迅速,然而疫苗质控方面仍缺乏统一的检测方法和标准物质,影响疫苗的研发进程。本文通过梳理mRNA疫苗质控现有国内外有限的指南文件和文献,总结归纳疫苗质控的考虑要点,以期为mRNA疫苗质量控制提供参考。
1 mRNA疫苗成分和生产工艺
mRNA疫苗由编码抗原的活性成分mRNA和辅料组成。辅料包括递送载体、缓冲液、糖类和分散剂[21]。脂质纳米颗粒(lipid nanoparticles,LNP)是最常用的递送系统,可包裹mRNA后递送到细胞内。为避免mRNA降解,mRNA-LNP通常低温储存和运输,其中糖类作为冷冻保护剂,可避免mRNALNP结构遭到破坏[21]。mRNA疫苗生产过程包括DNA模板的制备、mRNA原液制备、mRNA疫苗制剂及灌装3个阶段[22-24],其具体步骤(见图1):1)种子活化。从种子库中取出冻存的种子,活化后接种发酵。2)质粒大量提取纯化。大肠埃希菌大批量发酵培养、完成质粒的提取和纯化。3)DNA模板制备。通过对质粒进行酶切和DNA纯化获得线性DNA模板。4)mRNA的体外转录和加工。以线性DNA为模板,通过体外转录和转录后加工或共转录的方法获得大量mRNA转录产物。5)mRNA纯化。体外转录加工的产物经纯化后获得mRNA原液。6)mRNA-LNP制剂制备。采用冲击式射流混合法、微流控混合法等技术将mRNA原液和LNP按一定比例进行精确混合后形成粒径均一的包裹mRNA的纳米脂质体,经超滤换液等步骤,去除杂质后制成mRNA-LNP制剂中间产物。7)mRNA疫苗灌装。mRNA-LNP制剂中间产物通过质量检测后进行无菌灌装,获得mRNA疫苗成品[23-24]。
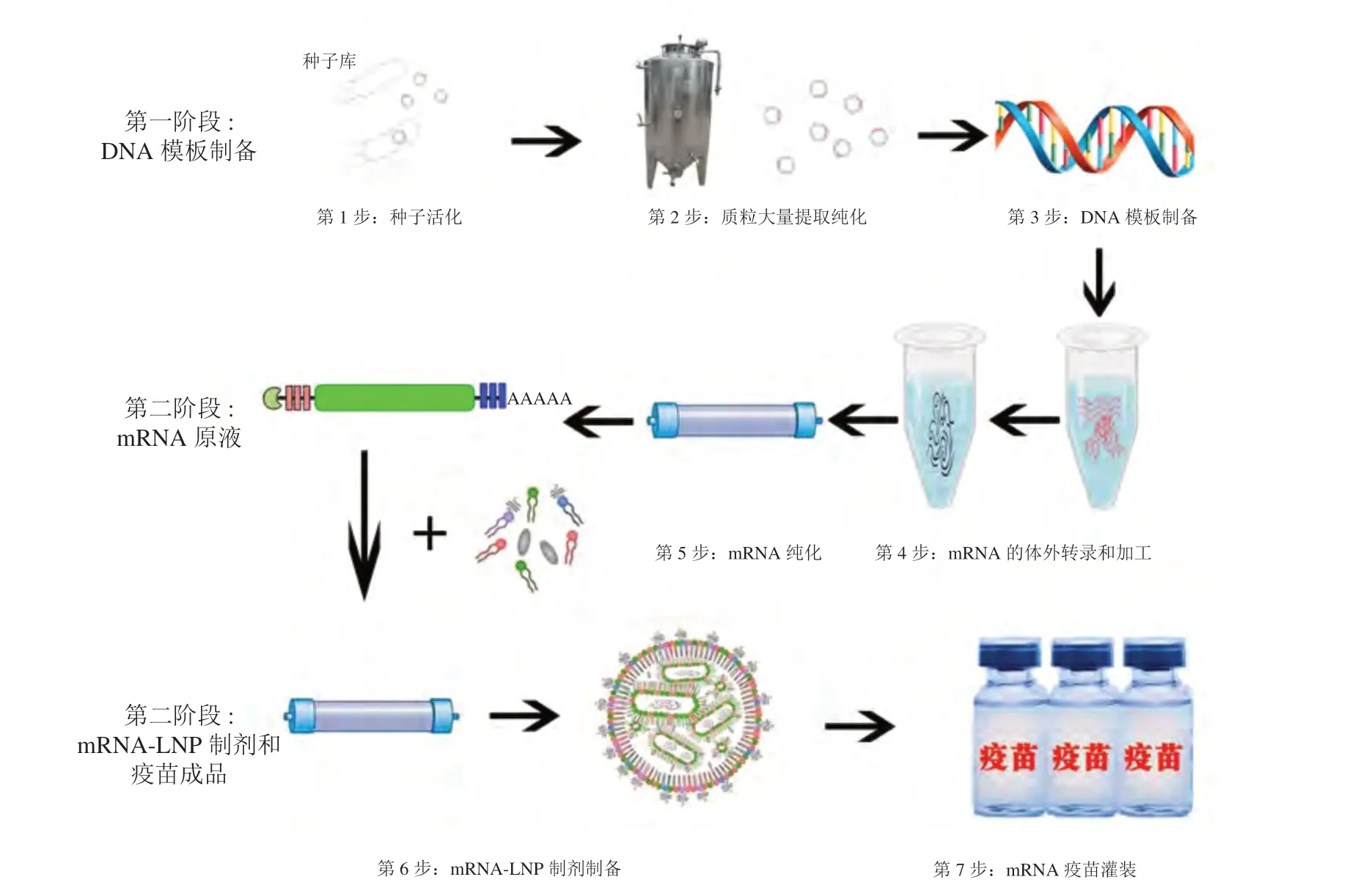
图1 mRNA疫苗生产工艺流程图Figure 1 Flow chart of mRNA vaccine manufacturing processes
2 mRNA疫苗质量控制
2.1 mRNA疫苗质量控制相关指南文件
2020年8月,国家药品监督管理局药品审评中心(Center for Drug Evaluation,CDE)发布了《新型冠状病毒预防性疫苗药学研究技术指导原则(试行)》,用于指导特殊应急状态下mRNA疫苗的研发[1]。2020年12月2日,辉瑞和BioNTech公司合作研发的mRNA新冠病毒疫苗BNT162b2在英国获批紧急使用,同月世界卫生组织(World Health Organization,WHO)发布了“评估基于RNA的传染病预防性疫苗的质量、安全性和有效性:监管考虑因素”的指南文件[25-26]。近期,美国药典委员会(United States Pharmacopeia,USP)为帮助研发者及企业免于将大量精力和资源用于自建检测方法,同时提升监管部门对企业疫苗进行有效质控的信心,颁布了针对疫苗鉴别、含量、完整性、纯度以及安全性和其他6类关键质量属性的通用检测方法和操作细则[27]。现有的各指南均基于目前mRNA疫苗公开发布的信息,许多候选mRNA疫苗的细节如检测项目以及分析方法和接受标准并未公开[28-29],这影响了mRNA疫苗质控的标准化和一致性研究,以及具体的国际指南制定[30-31]。
2.2 质控项目和标准制定
2.2.1 质控项目mRNA疫苗核酸结构和脂质体包裹制剂的特性决定了该类疫苗质控的特点。mRNA序列和完整性、含量及纯度、加帽率和包封率是疫苗特有的决定有效性和安全性的关键质量参数[31-33]。目前指南文件和文献报道疫苗质控项目主要包括如下几个方面。1)原材料质控:WHO指南要求对以DNA模板为主的原材料应进行检定和签发,应提供有关其来源、质量控制、稳定性等相关信息;要求确保包装材料不会对疫苗的质量产生不利影响,作为质量控制的一部分,需要对包装材料进行封闭完整性测试和可提取物和(或)可分离物的评估[26]。CDE指导原则要求如DNA模板制备涉及质粒构建及工程菌的使用应建立种子库系统,并需获得国家药检机构的检定报告。CDE要求提供工程菌以外的生产用其他原材料和辅料的来源、质量标准及检定报告,要求提供直接接触制品的包装材料和容器的来源、选择依据及质量标准等研究材料[1]。2)原液质控:除安全性指标外,mRNA疫苗原液阶段质控主要包括mRNA鉴定和杂质控制2个部分。mRNA鉴定包括外观、鉴别、pH值、序列长度、序列完整性及准确性、含量、加帽率、加尾结构或长度和纯度等指标。疫苗杂质包括产品相关杂质(如不完整mRNA、双链RNA等)和工艺相关杂质(如残留蛋白酶、DNA模板残留、金属离子残留等)。3)成品质控:成品阶段除常规的疫苗外观、装量、pH值、安全性和效力项目外,质控项目主要针对mRNA鉴定(鉴别、含量、纯度、完整性),递送系统各组分鉴定(鉴别、含量),制剂特性项目[包封率、纳米颗粒粒径、分散系数(PDI)、Zeta电位]以及工艺相关杂质残留(有机溶剂等)3个部分。
CDE建议在研发早期阶段检测疫苗体内效力,上市后通过足够批次的体内和体外效力数据及其与临床批次的对比分析,再评价体外效力代替体内效力的可行性[1]。WHO指南明确指出应考虑建立适当的体外替代方法来进行效力评价,在产品放行时建立体外方法和动物模型中效力的桥接是非常重要的。对安全性指标,CDE认为安全性指标通常包括异常毒性检查,而WHO则不要求进行异常毒性检查[1,26]。
2.2.2 质量标准疫苗质量标准研究时需从产品的设计目标着手,全面考虑安全性、有效性、工艺、检测方法、稳定性等内容,依据所使用检测方法总误差、多批次、临床批次和稳定性数据综合制定[34]。
WHO认为在研发早期质量标准可宽松,最终标准应基于临床试验中已被证明安全有效的批次检测结果制定,如疫苗效力标准应基于临床试验中证明疗效的最低剂量以及人类免疫原性数据设定,同时要求提供分析方法和接受限度的描述。CDE要求疫苗申报临床时可根据工艺确认资料,初步确定质量标准;上市阶段应按照相关指导原则进行风险控制分析,并结合工艺验证情况提供完整的质量标准[1,26]。
2.3 检测方法
2.3.1 检测方法建立和验证mRNA疫苗是全新的疫苗种类,检测方法是疫苗质控的基础;针对mRNA、脂质体和制剂的特性以及相关杂质等mRNA疫苗关键质量属性,研发企业应建立检测方法[35]。近期颁布的美国药典通则<1220>将生命周期管理理念应用到检测方法中,使方法设计、确认、转移和验证活动整合到检测方法生命周期过程中,并将其视为一个连续动态而非割裂的不同独立阶段的活动,以提升分析方法的质量水平[36]。ICHQ2(R2)及Q14征求意见稿进一步明确了对检测方法建立和验证的技术要求[37-38]。
CDE要求申报临床时提供的方法学验证资料应能初步证实检测方法的适用性,对关键质量属性(如包封率、加帽率、效力等)的检测方法,应提供验证与研发阶段控制及重要性相符或适用的资料,在上市阶段应提供全面的方法学验证资料。对产品安全性相关的质控指标(如微生物污染控制指标、有害物质残留等),建议尽早进行方法学验证,至少对适用性进行确认研究。WHO则要求检测方法应定义可接受限,并具有验证材料[1,26]。
2.3.2 参考检测方法对鉴别实验,WHO建议可选择直接测序、逆转录-PCR(reverse transcription-PCR,RT-PCR)测序和二代测序(next generation sequencing,NGS),检测纯度可采用凝胶电泳、毛细管电泳和(或)高效液相色谱(high performance liquid chromatography,HPLC);CDE建议鉴别实验方法除测序外,可选择电泳、HPLC等方法,而对含量检测(包括核酸浓度及包封率)以及产品相关杂质检测,可采用紫外光谱法或荧光分析,以及质谱、核磁及HPLC等适宜方法[1,26]。
美国USP提供了mRNA疫苗鉴别实验、完整性检测和纯度检测方法及操作细则,为研发企业统一疫苗关键质量参数的检测方法以及相应质量标准之间可比奠定了基础。USP规定mRNA疫苗鉴别实验检测方法为NGS、一代测序(sanger sequencing,桑格法测序)或RT-PCR;含量检测方法为逆转录定量PCR(reverse transcription quantitative PCR,RT-qPCR)和逆转录数字PCR(reverse transcription digital PCR,RT-dPCR),紫外光谱法(ultraviolet spectroscopy,UV);完整性检测包括完整mRNA和片段mRNA的百分比、5'加帽率、3'poly(A)加尾和mRNA完整性,完整mRNA和片段mRNA的百分比通过毛细管凝聚电泳法检测(capillary gel electrophoresis,CGE)、mRNA 5'加 帽率 通 过离子对反相高效液相色谱法检测(ion pair reversedphase high performance liquid chromatography,IPRP-HPLC)、3'poly(A)加尾通过反相高效液相色谱法检测(reversed-phase high performance liquid chromatography,RP-HPLC)、mRNA完整性通过凝胶电泳法检测(gel electrophoresis);纯度包括双链RNA(double strand RNA,dsRNA)和 DNA模板残留检测,分别采用免疫印迹法(immunoblot)和定量PCR(quantitative PCR,qPCR)法检测[27]。
近期BioNTech公司开发出一种体外转录mRNA加帽率测定方法,通过核酶在独特位置切割体外转录mRNA,释放出长约30 nt的5'加帽或未加帽切割产物,纯化后经变性胶或液相色谱-质谱法(liquid chromatography–mass spectrometry,LCMS)定量分析检测mRNA加帽率[39]。
2.4 疫苗标准物质
WHO要求企业建立疫苗内部参考品用于检测方法的标准化,在申请上市许可时提供参考品信息,选择充分表征结构、纯度和生物活性,经过临床试验评估的批次作为候选参考原料。在前期开发中可应用中试批的mRNA疫苗批次作为参考品,后期确定并表征适宜的临床试验批次用于商业批生产。为比较不同实验室和不同企业疫苗的检测结果,WHO已计划制备并提供以国际单位(international unit,IU)表示的国际标准品(international standard,IS)。目前WHO已建立新冠病毒抗体检测IS[40]。
CDE要求企业在申报临床阶段应建立核酸含量、纯度和生物活性等检测的参考品或对照品,提供参考品或对照品的来源、制备、检定结果、标定过程及稳定性研究等的初步研究资料。我国近期也完成新冠病毒中和抗体检测国家标准品的标定,可用于mRNA疫苗的体内效价检测方法质控以及临床评价[41]。
3 结语
全球新冠疫情极大加速了mRNA疫苗的研发和应用进程[42]。作为一种全新类型的疫苗,mRNA疫苗由于其可编辑、快速大量生产及较易质控等特点,迅速成为预防性和治疗性等疫苗研发的热点[43]。然而,该类疫苗的大规模生产和质控方面仍缺乏经验,相关的法规和指南文件也有待进一步完善和配套。我国进入临床的mRNA新冠病毒疫苗数量居世界前列,同时也启动了多个治疗性疫苗项目,但至今仍无疫苗批准上市。目前我国mRNA疫苗质控方面仍缺乏统一的检测方法和标准物质,不同研发企业的质控等信息难以共享,如何整合疫苗监管、研发和生产的资源,建立统一标准和质控体系,是目前我国mRNA疫苗行业发展面临的关键问题。企业应依据风险管理的理念设计、研发生产疫苗,科学合理设置质控项目和可接受标准,建立符合预期目的的简便、准确的检测方法。监管部门应推动质控标准的统一和标准化方法的应用,为推动mRNA疫苗发展,研发安全有效的疫苗提供基础。
猜你喜欢
今日农业(2021年2期)2021-11-27 19:19:53
中学生数理化·七年级数学人教版(2021年6期)2021-11-22 07:50:58
中学生数理化·七年级数学人教版(2021年6期)2021-11-22 07:50:58
中学生数理化·七年级数学人教版(2021年6期)2021-11-22 07:50:58
今日农业(2021年1期)2021-03-19 08:35:38
中国生殖健康(2020年5期)2021-01-18 02:59:40
疯狂英语·初中天地(2020年5期)2020-06-22 08:47:54
家教世界·创新阅读(2020年4期)2020-06-03 04:38:56
家教世界(2020年10期)2020-06-01 11:49:26
家教世界(2020年7期)2020-04-24 10:57:58
