
通过式固相萃取-超高效合相色谱-串联质谱法测定保健食品中19 种性激素
2022-10-31 08:56:58杨光勇梁秋艳
食品科学 2022年20期
杨光勇,梁秋艳,阳 胜,罗 岩,宋 敏,袁 辉
(新疆维吾尔自治区产品质量监督检验研究院,新疆 乌鲁木齐 830011)
性激素对于促进胎儿肝胆发育、维持人体正常的生理特性、调节人类的精神状态和认知能力等至关重要。此外,适量的性激素治疗对于延长癌症患者存活期具有积极作用。但过度摄入性激素则会导致儿童性早熟、人体内分泌紊乱,甚至危害生殖功能。随着国内人口老龄化以及国民保健意识的提高,保健食品逐步走入广大家庭。但是,近年相关研究显示,国内保健食品中性激素的非法添加状况不容乐观。因此,监测和评估保健食品中性激素的暴露风险对于保护消费者身心健康、维持正常的市场经济秩序等意义重大。
目前,性激素的检测方法有胶体金法、免疫分析法、毛细管电泳法,这些方法分别存在定性分析能力差、定量分析结果不可靠等缺点。液相色谱法、气相色谱-质谱法和液相色谱-质谱法也是分析性激素的常用方法。液相色谱法操作简单、应用广泛,但该方法定性能力不足,难以进行多组分同时分析。激素类化合物具有沸点高、挥发性低等特性,在使用气相色谱-质谱法进行检测时,需将性激素衍生为低沸点化合物,该操作不仅繁琐,还会影响定量结果的准确性。液相色谱-质谱法可有效弥补上述方法的不足,已成为检测性激素的重要方法之一,但该方法在进行正/负离子同时测定时,只能选择促离子化能力较为折中的流动相添加剂,无法最大程度地提高每一类性激素的质谱响应。
超高效合相色谱(ultra performance convergence chromatography,UPCC)以超临界CO为主要流动相,具有分离效率高、传质速率快等优点,较传统超临界流体色谱更易操作,已被成功应用于糖皮质激素、神经酰胺等多种生理活性化合物的分析。UPCC在性激素检测领域的应用较少,庞道标等采用UPCC-二极管阵列检测器对化妆品中违法添加的6 种性激素进行检测,4 min内即可实现基线分离,方法快速准确,但定性能力有待提高;Toit等采用UPCC-串联质谱测定了人体组织和血液中的11-羟基睾酮等3 种性激素,获得满意的分析结果;Quanson等采用UPCC-串联质谱对细胞培养液中的11-酮二氢睾酮等多种雄激素进行了分析,与反相超高效液相色谱-串联质谱法相比,灵敏度提高了5~50 倍,该方法灵敏度高、分离效果好,但需要使用多种氘代内标物质,这些内标物质价格昂贵且难以获取,限制了方法的进一步推广。本实验采用通过式固相萃取法对样品进行净化,以期建立UPCC-串联质谱测定保健食品中19 种性激素含量的方法。
1 材料与方法
1.1 材料与试剂
VE软胶囊、胶原蛋白粉、蛋白粉均为客户委托样品。
19 种性激素标准品(纯度≥96.3%)购自加拿大Toronto Research Chemicals公司、德国Dr.Ehrenstorfer公司、美国Sigma-Aldrich公司、美国Steraloids公司;正己烷、甲醇、乙腈(均为色谱纯) 美国Thermo Fisher公司;氨水(=25%~28%)、甲酸(优级纯) 美国Sigma-Aldrich公司;层析硅胶(200~300 目) 国药集团化学试剂有限公司。
1.2 仪器与设备
UPCC-TQS UPCC-三重四极杆质谱仪(配有电喷雾离子源(electrospray ionization,ESI)及MassLynx V 4.1 数据处理系统)、PRiME HLB 固相萃取柱(150 mg/3 mL) 美国Waters公司;Syncore Analyst平行蒸发定量浓缩仪 瑞士Büchi公司。
1.3 方法
1.3.1 标准品溶液的制备
称取适量各性激素标准品,用甲醇溶解并稀释成质量浓度为1 g/L的单标储备液;再准确吸取适量各单标储备液,用甲醇稀释成质量浓度为1 mg/L的混合标准使用液。将上述溶液置于-22 ℃冰箱保存。
1.3.2 样品溶液的制备
将软胶囊剖开,取油状内容物充分混匀;蛋白粉样品直接取用。称取2 g(精确至0.001 g)样品至15 mL具塞玻璃离心管中。在装有油状样品的离心管中加入10 mL正己烷,涡旋使样品溶解,加入2.0 g硅胶粉,涡旋混合2 min,5 000 r/min离心5 min,弃去正己烷溶液,在沉淀物中加入10 mL甲醇,涡旋混合2 min,5 000 r/min离心5 min,取甲醇溶液待净化。在装有固体样品的离心管中加入少量纯水,静置15 min使样品充分润湿,加入10 mL甲醇(含体积分数1%甲酸),涡旋混匀1 min,超声提取15 min,加入1.0 g无水硫酸镁,涡旋混合1 min,5 000 r/min离心5 min,取甲醇溶液待净化。将5 mL甲醇提取液转移至PRiME HLB固相萃取柱(150 mg/3 mL),收集流出液,待溶液流尽后加入4 mL乙腈洗脱,收集、合并流出液和洗脱液,45 ℃真空定量浓缩至1 mL,过0.22 µm滤膜。
1.3.3 仪器条件
(2)合理设置施工段数。以满足正常的专业工种能力为基础,施工段数如果设置过多,施工人员和施工设施会有所增加,施工难度也会有所增加,阻碍正常的施工组织开展。施工段数如果设置过少,也会对整个工程的进度产生严重的影响。
色谱条件:UPCC Torus 2-PIC 色谱柱(3.0 mm×100 mm,1.7 µm);柱温为45 ℃;流动相:A为超临界CO,B为甲醇(含体积分数0.1%甲酸);流速为1.25 mL/min。洗脱梯度程序:0~2.0 min,97.5% A、2.5% B;2.0~8.0 min,97.5%~88% A、2.5%~12% B;9.0~10.0 min,88%~97.5% A、12%~2.5% B。系统背压13.79 MPa;补偿液C和D分别为97%甲醇溶液(含体积分数0.2%甲酸)、98%乙腈溶液(含体积分数0.2%氨水),补偿液切换时间为1.9~2.4 min,100% D,5.0~5.5 min,100% D,其余时间段为100% C;补偿液流速为0.3 mL/min;进样体积为2 μL。
质谱参数:ESI源,雄激素、孕激素采用正离子电离模式,雌激素采用负离子电离模式;采集方式为多反应监测(multiple reaction monitoring,MRM)模式;离子源温度150 ℃;去溶剂气流量750 L/h;去溶剂气温度400 ℃;毛细管电压3.0 kV。
19 种性激素的基本信息见表1。
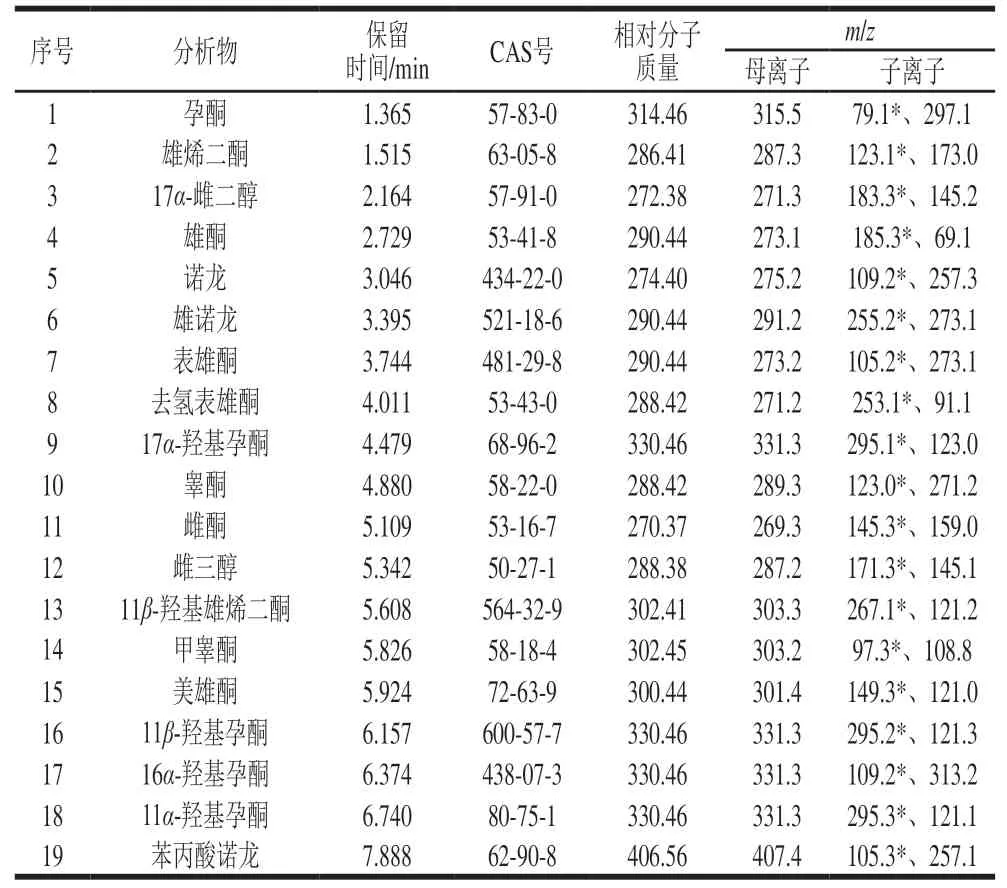
表1 19 种性激素的保留时间、CAS号、相对分子质量、母离子及子离子Table 1 Retention times,CAS numbers,relative molecular masses (Mr),parent ions,daughter ions of 19 sex hormones
2 结果与分析
2.1 色谱和质谱条件的优化
2.1.1 色谱柱的选择
19 种性激素均为环戊烷多氢菲衍生物,其中有4 组共12 种化合物互为同分异构体,甚至差向异构体,实验需选用合适的色谱柱,以实现上述化合物的基线分离,避免具有相同/的母离子和子离子相互干扰。为此,实验分别考察Torus 2-PIC(3.0 mm×100 mm,1.7 µm)、HSS CSB(2.1 mm×150 mm,1.8 µm)、BEH(3.0 mm×100 mm,1.8 µm)3 种色谱柱对分离效果的影响。结果表明,选用HSS CSB色谱柱和BEH色谱柱均无法实现11-羟孕酮和11-羟孕酮、雄酮和表雄酮等差向异构体的基线分离;选用Torus 2-PIC色谱柱时,各目标化合物分离效果最好,可能由于该色谱柱填料提供的偶极-偶极、电子转移、π-π等多重作用可有效识别出目标物空间结构的微小差异。但选用Torus 2-PIC色谱柱时,雌激素色谱峰略有拖尾,仍需进一步优化其他色谱条件。
2.1.2 色谱柱温度及系统背压的选择
超临界CO的洗脱能力会随色谱柱温度和系统背压的改变而变化,当柱温升高、背压减小时,流动相密度变小,洗脱能力降低,化合物保留时间延长。实验分别考察不同柱温(25、35、45、55 ℃)、不同背压(12.07、13.10、13.79、14.48 MPa)对分离效果的影响。结果表明,化合物的分离效果并未随背压的改变而出现明显变化;柱温升高时,分离趋势增加,当柱温超过45 ℃后,分离度反而减小,可能因为升高柱温加剧了分子的热运动,从而改变了化合物在色谱柱中的保留特性。综合考虑分离度、系统压力极限等因素,选取45 ℃色谱柱温度、13.79 MPa系统背压为测定条件。
2.1.3 助溶剂的选择
实验在优化的色谱条件下对19 种性激素混合标准溶液(20 µg/L)进行测定,其提取离子流色谱图见图1。经计算,图1中个别性激素色谱峰的理论塔板数高达10,由此表明,UPCC具有出色的分离效率。

图1 19 种性激素混合标准溶液(20 μg/L)的提取离子流色谱图Fig.1 Extracted ion current chromatograms of mixed standard solution of 19 sex hormones
2.1.4 质谱采集参数的确定
将稀释后的标准品储备溶液直接注入离子源,分别在正、负两种电离模式下优化质谱条件。结果表明,在正离子模式下,雄激素和孕激素分子结构中的羰基、共轭双键等结构更易于获得氢离子,从而生成较高丰度的[M+H]母离子;雄酮、表雄酮、脱氢表雄酮在获得氢离子后,经过开环、重排等复杂的反应,使[M+HHO]母离子丰度高于其他加合物形式;在负离子模式下,雌激素则更易于失去酚羟基上的氢,生成较高丰度的[M-H]母离子。确定母离子后,开启IntelliStart自动调谐功能,采用自动调谐所得的多反应监测参数。
2.1.5 补偿液组成及流速的优化
在ESI源的金属毛细管中,超临界CO因失去压力控制而转化为气态,从而失去溶解能力。为使分析物能够顺利传输至离子源并发生电离,需在色谱柱后端引入适宜的补偿液。补偿液不参与色谱分离,但其组成和流速对化合物的色谱峰形和离子化效率具有重要影响。Sun Ping等的研究表明,使用甲酸酸化后的质子性溶剂作为补偿液更有利于提高分析物在正离子模式下的质谱响应,且流动相pH值至少低于样品p值2 个单位;使用碱化后的非质子性溶剂作为补偿液更有利于待测物[M-H]分子离子峰的形成,且流动相pH值至少高于样品p值2 个单位。因此,实验分别考察不同体积分数97%甲醇溶液(含体积分数0.1%、0.2%、0.3%甲酸)和不同体积分数98%乙腈溶液(含体积分数0.1%、0.2%、0.3%氨水)作为补偿液,以及补偿液在不同流速(0.1、0.2、0.3、0.4 mL/min)时对测定结果的影响。结果表明,选用97%甲醇溶液(含体积分数0.2%甲酸)和98%乙腈溶液(含体积分数0.2%氨水)分别作为正、负离子模式下的补偿液,可获得较好的色谱峰形和响应强度;补偿液流速为0.3 mL/min时,各性激素响应值最高(图2a);继续增加流速,则系统压力超过设定值,迫使背压阀开启排放功能,分流了部分性激素,使质谱响应值降低。

图2 补偿液流速(a)和补偿液中纯水体积分数(b)对3 种性激素峰高的影响Fig.2 Effects of flow rate (a) and water volume fraction (b) of compensation solution on the peak heights of three sex hormone
此外,在补偿液中加入适量纯水,可改变离子源中的气氛,有助于提高化合物的质谱响应。通过比较补偿液中添加不同体积分数(1%、2%、3%、4%、5%、6%)纯水时化合物的峰高(图2b),可知正、负离子模式下纯水的最佳体积分数分别为3%和2%;当超过5%后,系统压力波动超过仪器告警阈值,仪器始终无法进入就绪状态,可能因为此时流动相管路内难以形成稳定的超临界流体。
2.2 样品提取和净化条件的优化
性激素极性较弱,可选用甲醇、乙腈、丙酮等作为提取液。实验参考饶雅琨等的研究,选用甲醇提取固体样品中的性激素。李双等研究表明,在蛋白粉中加入适量纯水,可使样品产生溶胀,提高提取液的穿透性;酸性环境能有效降低雌激素在水中的溶解度,从而提高有机溶剂的提取效率。因此,实验以蛋白粉阴性加标样品(20 µg/kg)为对象,考察直接提取和加水润湿后提取、提取液中含不同体积分数(0%、1%、2%,图3a)甲酸,以及不同超声提取时间(5、10、15、20 min,图3b)对回收率的影响。结果显示,将样品润湿后,以1%甲酸-甲醇溶液超声提取15 min时各性激素回收率最高。

图3 不同体积分数甲酸(a)和超声提取时间(b)对19 种性激素回收率的影响Fig.3 Effects of different volume fractions of formic acid (a) and ultrasonic extraction time (b) on recoveries of 19 sex hormone
一些研究采用凝胶制备色谱法提取和净化油状样品中的性激素,但该方法效率低下,成本高昂且污染严重。因此,实验改进了黄百芬等的研究,先将样品溶解于正己烷中,再用硅胶粉末吸附溶液中的性激素,甘油酯、VE等非极性杂质则随正己烷一同弃去,最后用甲醇洗脱硅胶上吸附的性激素。该方法可除去样品中的大部分杂质,降低后续净化的难度,并且操作简单、成本低廉。实验以19 种性激素的正己烷溶液(10.0 µg/L)为模拟物进行验证,结果显示,经硅胶粉末吸附后的正己烷溶液中未检出性激素,而甲醇洗脱液中各性激素质量浓度均不小于9.7 µg/L。
以上样品提取液中仍含有大量杂质,需采取有效的净化手段进一步减小基质干扰。为提高分析通量,实验采用通过式固相萃取法净化样品提取液。张再永等的研究表明,PRiME HLB通过式反相固相萃取柱能有效保留甘油酯、磷脂、蛋白质等化合物,且使用前无需活化和平衡,可极大的提高工作效率。甲醇、乙腈均是反相固相萃取法常用的洗脱液,乙腈的洗脱能力更强,有利于减少试剂消耗并缩短浓缩时间。因此,实验以PRiME HLB固相萃取柱为净化柱,乙腈为洗脱液净化样品提取液。通过比较不同洗脱体积(0、1、2、3、4、5 mL)下各性激素的回收率可知,最佳洗脱体积为4 mL;超过4 mL后,回收率不再变化,但一级质谱扫描图谱中杂质峰簇明显增多。因此,实验最终采用4 mL乙腈进行洗脱。
2.3 方法学验证结果
2.3.1 基质效应
实验配制并测定了系列基质匹配标准溶液和系列空白溶剂标准溶液,基质效应=基质匹配标准曲线斜率/空白溶剂标准曲线斜率-1。结果表明,3 种样品的基质效应均在-0.51~0.20范围内,其中蛋白粉基质对雌激素具有较强的基质抑制效应,可能是在离子化过程中,溶液中残留的蛋白质、多肽等强极性杂质与雌激素共同竞争液滴表面,导致雌激素离子化效率降低;VE软胶囊基质中,各性激素的基质效应普遍较弱,这与张虹艳等的研究结论一致。为减小基质效应对定量分析准确性的影响,实验采用空白基质匹配标准溶液绘制标准曲线。
2.3.2 线性范围、检出限(limits of detection,LOD)和定量限(limits of quantitation,LOQ)
以阴性样品提取液配制各性激素质量浓度均为0.10~100 µg/L的基质匹配标准溶液,采用本方法进行分析,以目标物定量离子峰面积为纵坐标,对应质量浓度为横坐标绘制标准曲线。结果表明,19 种性激素在各自的质量浓度范围内线性关系良好,相关系数()不小于0.997 1(表2)。再以各目标化合物3 倍信噪比(=3)确定LOD,以10 倍信噪比(=10)确定LOQ。如表2所示,各性激素的LOD和LOQ分别为0.03~0.85 µg/kg和0.10~2.5 µg/kg,与Toit、Quanson等报道的数值水平相当,优于李双、饶雅琨、罗文静等的报道。
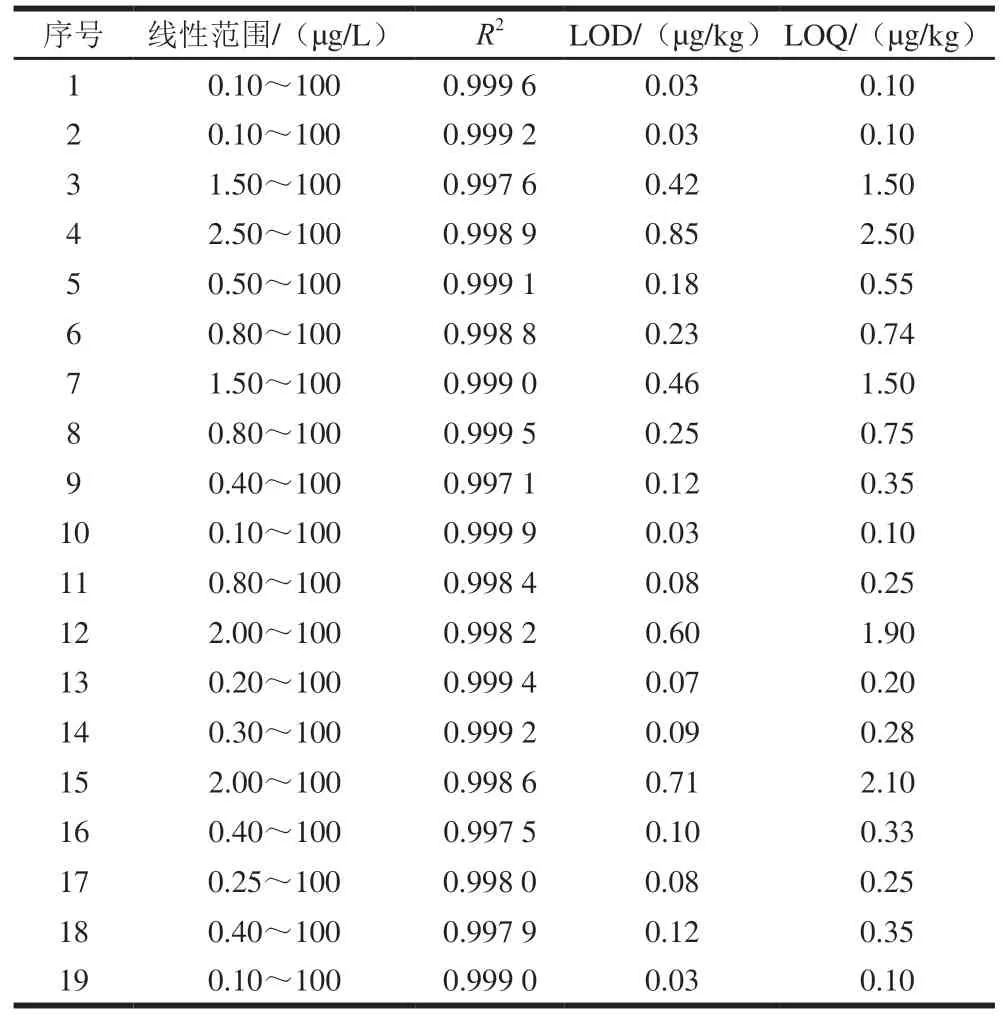
表2 19 种性激素的线性范围、线性方程、R2、LOD及LOQTable 2 Linear ranges,R2,LODs,and LOQs of 19 sex hormones
2.3.3 回收率、精密度实验结果
以阴性样品进行加标回收实验,加标量分别为0.5、2 mg/kg和10 mg/kg,每个加标水平制备3 份,按1.3.1和1.3.2节方法处理后进行测定,得到样品的平均回收率(=9)为77.8%~111.7%,相对标准偏差(relative standard deviation,RSD)为1.1%~8.4%(表3)。与李双、饶雅琨、罗文静等的研究相比,回收率和精密度均能满足保健食品中性激素非法添加检测的要求。

表3 阴性样品的加标回收率和RSDTable 3 Recoveries of spiked blank samples and precision RSDs
2.3.4 实际样品的测定
按照建立的方法对2 份VE软胶囊样品、2 份胶原蛋白粉样品和4 份蛋白粉样品进行测定。其中1 份胶原蛋白粉样品中检出孕酮和雌三醇,含量分别为1.42、0.88 mg/kg;1 份蛋白粉样品检出诺龙、睾酮和雌三醇,含量分别为11.05、2.21 mg/kg和0.40 mg/kg;1 份蛋白粉样品检出诺龙、睾酮和甲睾酮(图4),含量分别为7.39、1.53 mg/kg和0.59 mg/kg,其他样品均未检出这19 种性激素。蛋白粉类样品的总体检出率为50%,与李双等的研究基本一致。
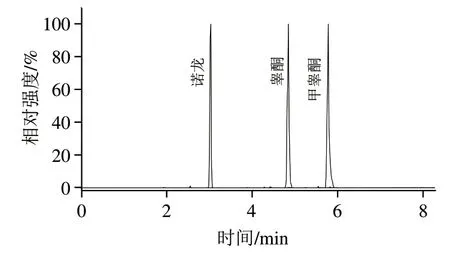
图4 阳性蛋白粉样品(6号样品)的总离子流色谱图Fig.4 Total ion current chromatograms of positive protein powder sample
3 结论
建立UPCC-串联质谱测定保健食品中19 种性激素含量的方法,采用优化的样品前处理方式,有效减小了基质干扰,并提高了检测灵敏度。方法快速准确、灵敏度高、专属性好、绿色环保,可在8 min内有效分离包括2 组4 种差向异构体在内的4 组共12 种同分异构体。采用建立的方法对实际样品进行检测,结果能满足一般化学分析的要求,对于保健食品中性激素的测定和暴露风险评估提供了可靠的技术支持。
猜你喜欢
现代临床医学(2024年1期)2024-01-30 12:40:24
煤化工(2022年3期)2022-07-08 07:24:42
农村百事通(2021年31期)2021-12-13 03:02:33
中老年保健(2021年3期)2021-08-22 06:51:08
中国生殖健康(2020年4期)2021-01-18 02:58:00
农村百事通(2021年11期)2021-01-17 07:33:01
中国生殖健康(2018年4期)2018-11-06 07:12:08
饮食保健(2016年21期)2016-12-10 05:28:39
中国资源综合利用(2016年10期)2016-01-22 08:36:09
湖南中医药大学学报(2015年1期)2016-01-06 01:06:39
