
加热温度对含蜡原油胶凝过程微观机理的影响
2022-10-21 01:16国丽萍季军美张家俊张利利贺怀东施少军
东北石油大学学报 2022年4期
国丽萍,季军美,2,张家俊,张利利,贺怀东,施少军,杨 宝
(1.东北石油大学 提高油气采收率教育部重点实验室,黑龙江 大庆 163318; 2.中国石化重庆页岩气有限公司,重庆 408400; 3.大庆油田有限责任公司 第二采油厂,黑龙江 大庆 163414; 4.大庆油田有限责任公司 第三采油厂,黑龙江 大庆 163113; 5.吉林油田有限责任公司 勘探开发研究院,吉林 松原 138000)
0 引言
易凝高黏的高含蜡原油在中国所产原油的占比超过80%,大多数含蜡原油具有凝点温度高的特点[1]。加热输送是中国高含蜡原油普遍采用的输送方式,由于事故或设备检修等导致管道停输,管道内原油的温度若降至析蜡点温度以下,蜡晶就开始析出。随温度的降低,析出的蜡晶聚集、交联而形成空间网状结构,原油胶凝并失去流动性,易造成凝管等灾难性事故[2]。含蜡原油的胶凝过程与经历的热历史有关[3]。为了保证易凝高黏原油管道输送安全、经济运行,需要提供可靠的基础数据,有必要研究含蜡原油体系胶凝过程的作用机制与微观机理。
目前,人们从微观分子层面对含蜡原油流变特性进行探讨。SHAHRUDDIN S等[4]建立长链正构烷烃的分子动力学模型,研究含蜡原油初始凝固状态时的相变化,较重的组分先被限制运动,较轻的组分保持流动性。CHEN Xuejiao等[5-6]采用分子动力学模拟方法,研究磁场对含蜡原油流变特性的影响,黏度降低主要集中在弱磁场强度范围内;考虑直流电场对含蜡原油黏度的影响,建立分子动力学模型,电场强度越大,黏度下降的幅度越大,温度越低,电降黏效果越好。在有关研究成果中,未见采用分子动力学研究加热温度对含蜡原油胶凝过程影响的报道。
根据油品实验基本物性资料,选取与实验油样实际组分结构较接近的蜡晶、胶质和沥青质分子模型,应用Materials Studio(MS)软件构建含蜡原油体系模型,研究不同加热温度对含蜡原油凝点变化的影响规律,以回转半径、分子间作用力、径向分布函数和均方位移为特征量,分析体系内部胶凝过程的微观机理,为热历史对含蜡原油胶凝体系流变特性的影响规律研究提供依据。
1 含蜡原油体系模型
1.1 模型构建
采用MS软件对含蜡原油体系进行分子动力学模拟。根据含蜡原油样品基本物性资料[7](见表1),建立含蜡原油体系模型,选取正构烷烃C22H46模拟蜡晶分子、Modified R-benzo-thio-S分子模型[8]模拟胶质分子、At-N+2S+O分子模型[9]模拟沥青质分子,作为含蜡原油体系模型构建基本单元(见图1)。蜡晶、胶质、沥青质的实际分子数通过质量分数与平均相对分子质量乘积获得,为研究同种分子间的微观作用机理,必须保证含蜡原油体系中每种分子至少存在2个,选取沥青质分子数为2、胶质分子数为14、蜡晶分子数为44。

表1 含蜡原油样品基本物性Table 1 Basic physical properties of waxy crude oil samples
1.2 模拟方法
根据基本单元以随机方式构建含蜡原油体系的初始结构模型,以能量最小化方式优化模型结构。采用Dynamics模块模拟计算凝点实验的降温过程,计算过程选择等温等压(NPT)系综、COMPASS力场,分别采用Ewald法和Atom Based法计算静电力和范德华力。模拟加热温度分别为30、40、50、60、70 ℃,降温速率为1 ℃/min。
在分子动力学模拟计算中,体系结构的合理性和稳定性可以通过计算过程中温度、能量的变化情况判断[10],体系的温度、能量在10%内波动时,体系达到平衡状态[11]。当加热温度为30 ℃时,模拟计算温度和能量随时间变化曲线见图2。能量在10%范围内波动,温度的波动区间以开氏温度计算。加热温度为30 ℃(303.0 K)时,温度在10%内的波动区间为272.7~333.3 K,将摄氏温度波动区间24~36 ℃换算成开氏温度区间为297.0~309.0 K,满足温度在10%内波动。此时,整个油样体系达到平衡,说明设定的模拟条件及模拟流程较合理。
2 模拟结果讨论
根据基本单元随机构建含蜡原油体系结构模型,分析体系达到平衡时的密度、自扩散系数、回转半径、分子间作用力、径向分布函数和均方位移等参数的变化规律,分析含蜡原油胶凝过程的微观作用机理。
2.1 凝点判断
含蜡原油体系的密度和自扩散系数随温度的变化曲线存在异变区间,由异变区间可判断凝点区间,根据两类曲线区间的交集界定模拟计算的凝点[12]。
2.1.1 密度分析
加热温度为40 ℃时,含蜡原油体系密度随温度(温度步长为5 ℃)的变化曲线见图3(a)。由图3(a)可知,随温度的下降,密度呈增加趋势。在模拟原油凝点附近的温度区间,密度发生突变,根据密度—温度曲线转折点可判断含蜡原油体系的凝点区间[13]。密度曲线的异变区间为10~20 ℃,即油样的凝点区间。
2.1.2 自扩散系数分析
自扩散系数是反映含蜡原油体系中分子迁移和活性的重要特征量,随温度的降低,自扩散系数逐渐减小[14]。这是由于含蜡原油体系在接近凝点温度时,阻碍分子运动,特别是长链型分子的迁移和活跃度急剧降低,使自扩散系数急剧减小。自扩散系数发生突变的温度点认为是体系的相变点,可以判断含蜡原油体系的凝点区间。加热温度为40 ℃时,含蜡原油体系自扩散系数随温度(温度步长为5 ℃)的变化曲线见图3(b)。由图3(b)可知,自扩散系数曲线的异变区间为15~25 ℃,即油样的凝点区间。
2.1.3 综合判断
对于加热温度为40 ℃的含蜡原油体系,由密度和自扩散系数曲线判定油样凝点出现的温度区间为15~20 ℃。为了更准确获得油样的凝点温度,在凝点出现的温度区间以1 ℃的温度步长进行模拟计算,分别绘制密度、自扩散系数随温度变化曲线(见图4)。由图4可知,密度的突变温度为16 ℃,自扩散系数的突变温度为19 ℃。自扩散系数的突变温度高于密度的,这是由于在含蜡原油体系中,当自扩散系数发生突变时,局部分子的迁移性下降没有对整个含蜡原油体系产生影响,在油样中存在析蜡结晶,但总体上没有失去流动性[15]。选择密度的突变温度作为模拟计算的凝点温度,在加热温度为40 ℃时,含蜡原油体系的凝点温度为16 ℃。
2.2 凝点变化
为研究加热温度对含蜡原油凝点的影响,根据凝点判断方法,模拟5个加热温度下的密度、自扩散系数的异变区间及其交集,缩小模拟计算的温度步长,根据密度的突变温度,判断不同加热温度下含蜡原油体系的模拟凝点温度,并与文献[7]实验数据进行对比。不同加热温度下,模拟凝点和实验凝点绝对误差最大为2 ℃(见表2)。文献[7]实验结果根据SY/T 0541—1994《原油凝点测定法》测定,测量误差为1~2 ℃。因此,可以认为含蜡原油凝点温度的模拟结果可信,说明含蜡原油分子体系的正确性和模拟方法的可靠性。
加热温度对含蜡原油的胶凝过程有恶化影响[16]。含蜡原油凝点随加热温度变化曲线见图5,其中实验数据来自文献[7]。由图5可知,分子动力学模拟计算的凝点恶化加热温度为50 ℃,与实验数据相吻合。
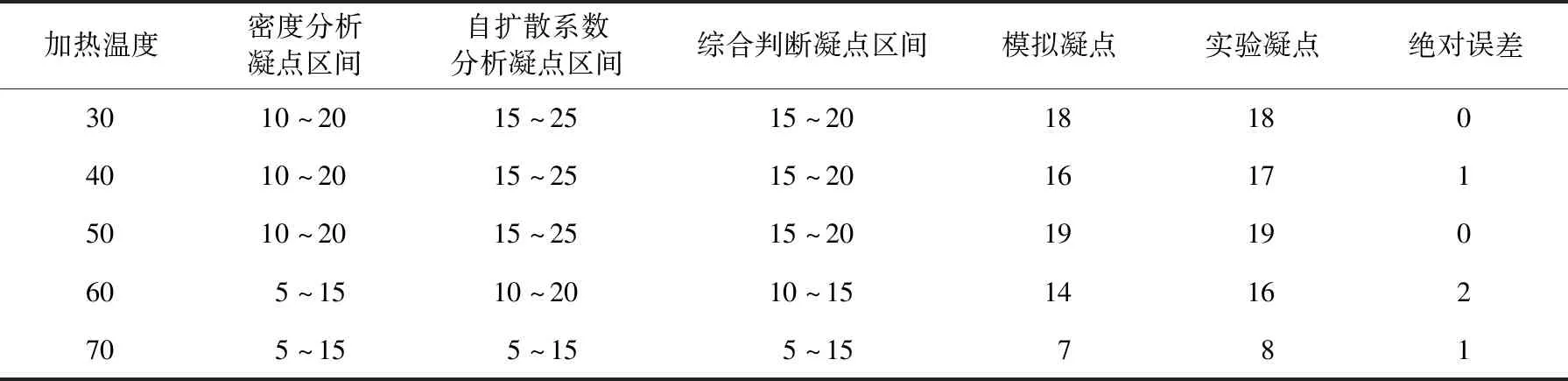
表2 不同加热温度下判断含蜡原油凝点相关数据Table 2 Data sheet related to condensation point of waxy crude oil system at different heating temperatures ℃
2.3 微观机理分析
2.3.1 回转半径
在凝点温度附近含蜡原油胶凝体系中分子链发生拉伸与扭转[17]。回转半径(Rg)为分子动力学中描述分子链构型的参量,其表达式为
(1)
式中:rx为第x个原子的位置矢量;ry为分子链质心的位置矢量;m为分子链上的原子数。
不同加热温度下,降温过程中含蜡原油体系回转半径概率分布见图6。由图6可知,不同加热温度下,含蜡原油体系回转半径的概率近似于高斯分布;随温度的降低,体系的回转半径整体向左偏移;在凝点(见表2)附近,回转半径向左出现相对大幅偏移,表明随温度的降低,分子链的构象由最初的直链逐渐转变成卷曲状态,且在凝点附近变化最为明显。
2.3.2 分子间作用力
对于平衡状态的含蜡原油体系,采用MS软件可以模拟计算同种和异种分子体系的分子间相互作用能,异种分子间的相互作用能[18]表达式为
EM-N=ET-EM-M-EM-N,
(2)
式中:ET为M、N分子共同存在时体系总分子间相互作用能;EM-M、EN-N分别为体系中M、N分子间的相互作用能。
模拟计算3种同种与3种异种分子间相互作用能(见表3)。分子间作用能为负值,表示分子间相互作用力为吸引力[19]。分子间相互作用能绝对值越大,分子间相互作用越强。由表3可知,蜡晶分子间的相互作用能明显高于其他分子间的,与宏观流变实验研究认为蜡晶的存在是含蜡原油流变性复杂的根本原因相吻合。

表3 含蜡原油体系中分子间相互作用能Table 3 Intermolecular interaction energy in waxy crude oil system
为研究分子间各种作用力的贡献,采用MS软件模拟计算分子间内聚能(使分子在体系中彼此分离到无穷远处需要的平均能量)密度,内聚能密度(CED)是指体系单位体积的内聚能,是评价分子间作用力大小的物理量[20-21]。在模型的计算力场中,将内聚能密度视为克服分子间非键合力需要的能量,也是克服范德华力和静电力需要的能量之和[22]。模拟计算蜡晶、胶质、沥青质3种分子模型内聚能密度(见图7)。由图7可知,蜡晶分子总内聚能密度明显大于其他两种分子的,说明蜡晶分子间相互作用最强,蜡晶分子是决定含蜡原油胶凝结构形成的主要因素。蜡晶分子间相互作用力主要是范德华力,占比为96.7%,静电力占比为3.3%,因为蜡晶分子由烷烃构成;胶质分子间相互作用力主要是范德华力,占比为93.9%,静电力占比为6.1%,因为胶质分子是含芳香环的多支链大分子;沥青质分子间相互作用力中范德华力占比为82.7%,静电力占比为17.3%,相比前两种分子,沥青质分子的静电力占比增加2.8~5.2倍,因为沥青质分子除C、H外,还含有O、N等原子,易形成O—H、N—H键。
2.3.3 径向分布函数
径向分布函数(RDF)是指区域内单位体积的粒子数与体系平均单位体积的粒子数之比,能够反映流动过程中分子聚集的程度[23],其表达式为
(3)
式中:ρ为体系粒子的密度;N为体系中粒子数;dN为半径r、厚度dr的球壳内粒子数。
不同加热温度下,降温过程中蜡晶—蜡晶、蜡晶—胶质和蜡晶—沥青质分子径向分布函数见图8-10。由图8-10可知,蜡晶—蜡晶分子间的RDF出现峰值且集中,RDF峰值发生在半径为0.111 nm处,RDF集中的半径区间为0~0.500 nm,说明同种分子主要聚集于此[24-25];蜡晶—胶质、蜡晶—沥青质异种分子间的RDF在不同位置发生不同程度的聚集,RDF大多出现在半径大于0.500 nm处;同种分子的RDF峰值远大于异种分子的,表明同种分子的聚集更为集中,蜡晶的聚集在含蜡原油的胶凝过程中起主导作用。
为进一步研究加热温度对含蜡原油胶凝过程的影响,模拟计算不同加热温度下,降温过程中蜡晶—蜡晶分子间径向分布函数峰值(g(r)max)(见图11)。由图11可知,加热温度不变时,蜡晶分子的g(r)max随温度的降低而升高,聚集程度增加,与宏观流变实验温度越低、胶凝结构越强的规律[16,26]相吻合。不同加热温度下,蜡晶分子的g(r)max在凝点温度(见表2)附近发生阶跃式增加(图中红色数据点)。加热温度为50 ℃时,g(r)max曲线明显高于其他加热温度的,与宏观流变实验结果吻合,为含蜡原油流变性存在凝点恶化加热温度的原因。
2.3.4 均方位移
均方位移(MSD)可以表征含蜡原油体系内分子运动状况,其表达式[27]为
MSD=〈|r(t)-r(0)|2〉,
(4)
式中:r(0)为分子的初始位置坐标;r(t)为分子在时间t的位置坐标。
模拟计算不同加热温度下,降温过程中含蜡原油体系蜡晶主链均方位移(见图12)。对于不同加热温度的含蜡原油体系,随模拟计算时间的延长,蜡晶分子的MSD在凝点温度附近存在区域分界(图中绿色曲线),出现两种变化趋势:高于凝点温度时,MSD随时间的延长而大幅增加,曲线以恒斜率上升,分子运动速度恒定;低于凝点温度时,MSD随时间的延长而小幅增加,曲线上升斜率逐渐减小,分子运动减慢,直至体系胶凝。
不同加热温度下蜡晶主链MSD区域分界线见图13。由图13可知,不同加热温度下曲线趋势一致,数值相差不大,表明处于胶凝状态的分子活性相似。
3 结论
(1)基于油品基本物性资料,选取与实验油样组分结构较接近的蜡晶、胶质和沥青质分子模型,采用Materials Studio软件构建含蜡原油体系模型,分析加热温度对含蜡原油凝点变化的影响规律,以及体系内部胶凝过程的微观机理。
(2)含蜡原油凝点可由密度—温度曲线和自扩散系数—温度曲线综合判断。随温度的降低,含蜡原油体系分子链的构象由直链逐步转变成卷曲状态,且在凝点附近变化最为明显。
(3)蜡晶同种分子间的聚集是含蜡原油胶凝的主要因素,范德华力是主要作用力。不同加热温度下,蜡晶同种分子间RDF存在峰值且集中在半径为0.111 nm处,凝点恶化加热温度的RDF峰值明显高于其他加热温度的,是含蜡原油流变性存在恶化加热温度的原因。
猜你喜欢
冰川冻土(2022年2期)2022-06-14
能源工程(2022年1期)2022-03-29
建材发展导向(2022年4期)2022-03-16
铁道建筑技术(2021年4期)2021-07-21
建材发展导向(2020年3期)2020-11-28
电脑知识与技术(2018年8期)2018-05-07
北京航空航天大学学报(2018年4期)2018-05-04
安徽农学通报(2017年17期)2017-09-21
科技视界(2016年19期)2017-05-18
科学与财富(2017年10期)2017-05-09
