
叔丁基对苯二酚对早期糖尿病模型大鼠视网膜结构和功能的保护作用及其机制
2022-10-16 13:32王娟韦芳曹阳田敏吕红彬
中华实验眼科杂志 2022年9期
王娟 韦芳 曹阳 田敏 吕红彬
1西南医科大学附属医院眼科,泸州 646000;2上海交通大学附属第一人民医院眼科 上海市眼底病重点实验室,上海 200080
糖尿病视网膜病变(diabetic retinopathy,DR)是糖尿病的一种常见微血管并发症,早期防治是防止DR致盲的重要手段之一[1]。研究表明糖尿病大鼠的视网膜神经功能损伤早于血管损伤[2]。临床研究也证实了糖尿病患者尚未出现视网膜结构改变时,其视网膜电图(electroretinogram,ERG)的b波及振荡电位已出现振幅下降,并且随着病程延长及病情加重,ERG振幅进一步下降,潜伏期延长[3],充分说明糖尿病患者早期视网膜神经功能即已出现异常。因此,早期保护和恢复DR中视网膜神经元的功能对于防止DR致盲有重要意义。叔丁基对苯二酚(tert-butylhydroquinone,tBHQ)分子式为C10H14O2,是一种合成的酚类抗氧化剂,常被用作食品添加剂以防止油脂食品氧化变质,属于Ⅱ相酶诱导剂之一,可有效防止氧化应激诱导的细胞功能障碍[4]。研究表明tBHQ可通过激活磷脂酰肌醇3激酶(phosphoinositide 3-kinase,PI3K)途径促进蛋白激酶B(protein kinase B,Akt)磷酸化[5];而磷酸化的Akt及其活化通路可对阿尔兹海默症等神经退行性疾病发挥抗细胞凋亡及抗氧化应激作用[6],证实了tBHQ在保护神经元方面具有一定作用。在DR中,PI3K/Akt途径受到抑制,并参与高糖诱导的视网膜细胞损伤[7]。因此恢复PI3K/Akt通路的活性将对防治DR起到关键作用。既往研究结果表明,tBHQ可通过促进Akt的下游靶蛋白内皮型一氧化氮合酶(endothelial nitric oxide synthase,eNOS)活化发挥抵抗内皮功能损害和eNOS解偶联的作用[8];而tBHQ能否恢复糖尿病诱导的eNOS活性尚未可知。本研究拟建立糖尿病动物模型和细胞高糖模型,观察tBHQ对DR早期视网膜结构和功能的作用,并探讨其作用机制。
1 材料与方法
1.1 材料
1.1.1实验动物 选取8周龄清洁级雄性SD大鼠45只,体质量180~200 g,购自上海西普尔-必凯实验动物有限公司。饲养环境为12 h/12 h循环明暗光照,温度为22~25 ℃,湿度为40%~70%。实验动物的饲养、使用与处死方法均遵循西南医科大学实验动物管理条例,研究方案经西南医科大学实验动物伦理审查委员会审查批准(批文号:201711189)。
1.1.2实验细胞 大鼠视网膜Müller细胞系rMC-1(美国西北大学Vijay Sarthy实验室)。将细胞培养于含体积分数10%胎牛血清(fetal bovine serum,FBS)的DMEM培养基中,培养环境为体积分数5% CO2、饱和湿度,温度为37 ℃。当细胞培养至80%~90%融合时,进行传代培养,取第3~8代细胞进行实验。
1.1.3主要试剂及仪器 tBHQ、链脲佐菌素(streptozocin,STZ)(美国Sigma公司);PI3K抑制剂LY294002(美国Merck公司);无糖DMEM培养基(美国Gibco公司);RIPA裂解液、蛋白上样缓冲液、超敏型增强化学发光(enhanced chemiluminescence,ECL)试剂、一抗及二抗稀释液(上海碧云天生物技术有限公司);小鼠抗大鼠eNOS抗体(ab76198,美国Abcam公司);兔抗大鼠p-eNOS ser1179抗体(12454-2,美国SAB公司);兔抗大鼠p-Akt ser473(4060s)、兔抗大鼠Akt(4685s)、β-actin、HRP标记单克隆抗体(5125s)(美国CST公司);TUNEL凋亡检测试剂盒(瑞士Roche公司)。Ganzfeld ERG System(美国Phoenix-Micron公司);NANO DROP分光光度计(美国Thermo Fisher Scientific公司);激光扫描共聚焦荧光显微镜(TCS SP8,德国Leica公司)。
1.2 方法
1.2.1动物分组及模型建立 将45只大鼠按照随机数字表法分为正常对照组、糖尿病模型组和tBHQ干预组,每组15只。适应性喂养1周后,糖尿病模型组和tBHQ干预组大鼠腹腔内注射质量分数1% STZ溶液(6.5 ml/kg)建立糖尿病大鼠模型,正常对照组大鼠腹腔内注射等容量枸橼酸钠缓冲液。正常对照组和糖尿病模型组大鼠喂食普通饲料(上海帆泊生物技术有限公司),tBHQ干预组自STZ注射前2周开始一直喂食质量分数1% tBHQ添加饲料(上海帆泊生物技术有限公司)。造模后72 h,大鼠尾静脉采血测量空腹血糖(fasting plasma glucose,FPG),以FPG>16.7 mmol/L为建模成功,以后每2周检测1次随机血糖并记录。
1.2.2细胞分组及模型建立 将Müller细胞系分为5个组,其中正常对照组细胞于含5.5 mmol/L葡萄糖的培养基中培养72 h;甘露醇对照组细胞于含5.5 mmol/L葡萄糖及24.5 mmol/L甘露醇的培养基中培养72 h;高糖组细胞于含30 mmol/L葡萄糖的培养基中培养72 h;tBHQ干预组细胞先于含5 μmol/L tBHQ的正常糖培养基中预处理24 h,然后于含5 μmol/L tBHQ的高糖培养基中继续培养72 h;PI3K抑制剂组细胞先于含5 μmol/L LY294002的正常糖培养基中预处理6 h,然后于含5 μmol/L LY294002及5 μmol/L tBHQ的正常糖培养基中继续培养24 h,最后于含5 μmol/L LY294002及5 μmol/L tBHQ的高糖培养基中继续培养72 h。每天更换培养基。实验重复5次。
1.2.3ERG检测大鼠视网膜功能 造模后4周,剔除发生糖尿病性白内障的大鼠,各组选取6只,暗适应至少12 h,使用质量分数10%水合氯醛腹腔内注射(3 ml/kg)麻醉,复方托吡卡胺滴眼液充分扩瞳,将大鼠俯卧位固定于实验台上,给予保温。采用Ganzfeld ERG系统记录暗适应视网膜反应。安装电极:角膜电极垂直于角膜顶点放置,充分接触角膜,使光线充分进入眼内;参考电极置于枕后矢状缝与两眼连线的交点处皮下;接地电极置于尾根部背侧皮下。根据前期预实验结果,刺激条件设置为:背景光强为0 cd/m2,闪光强度分别为-1.3、-0.3、0.7 log cd·s/m2,刺激间隔分别为30、40、50 s,单次刺激持续时间为10 μs,每个光强度记录5次得出平均值,换另一只眼再进行上述步骤。全部实验均在红光微弱照射下完成。记录数据,分析各组大鼠不同刺激光强度下ERG a波、b波振幅的差异。
1.2.4苏木精-伊红染色法观察视网膜组织形态结构 造模后4周,各组分别取5只大鼠,使用10%水合氯醛(3 ml/kg)腹腔内注射麻醉后并处死,取右眼置于质量分数4%多聚甲醛溶液中固定24 h。常规石蜡包埋,以平行于视神经方向行5 μm厚连续切片。取各组视网膜组织石蜡切片,依次放入二甲苯Ⅰ、Ⅱ和梯度乙醇中脱蜡水化,苏木素染色3~5 min,体积分数1%盐酸乙醇溶液浸泡5 s,体积分数1%氨水返蓝5 s,梯度乙醇脱水各5 min,体积分数0.5%伊红溶液复染5 min,无水乙醇脱水透明后中性树脂封片,光学显微镜下观察并拍照。
1.2.5TUNEL法观察视网膜组织细胞凋亡情况 造模后4周,各组分别取5只大鼠,使用10%水合氯醛(3 ml/kg)腹腔内注射麻醉并处死,取右眼置于4%多聚甲醛溶液中固定24 h,常规制作冰冻切片,以平行于视神经方向行12 μm厚连续切片。取各组视网膜冰冻切片室温风干,4%多聚甲醛室温固定20 min,磷酸盐缓冲液(phosphate buffer saline,PBS)清洗30 min,0.1% Triton X-100 4 ℃透膜2 min,PBS清洗,将配制好的TUNEL检测混合液滴于切片上,37 ℃孵育1 h;DAPI复染核并PBS洗涤后抗荧光淬灭封片剂封片。使用激光扫描共聚焦荧光显微镜观察并拍照,当红色荧光与细胞核重合时则该细胞为凋亡细胞,使用ImageJ软件分析整张视网膜图片,记录每张图片上的细胞总数及TUNEL染色阳性细胞数,凋亡指数=凋亡阳性细胞数/细胞总数×100%。
1.2.6Western blot法检测视网膜组织及细胞中p-Akt、Akt、eNOS、p-eNOS蛋白表达 取各组10只大鼠的左眼,沿角巩膜缘剪开去除晶状体,分离出视网膜组织,同时收集各组细胞,冻存于-80 ℃冰箱。利用RIPA裂解液提取视网膜组织和细胞内蛋白质,采用NANO DROP软件检测蛋白质浓度,并加入上样缓冲液煮沸后制备蛋白质样本。取各组蛋白样本30 μg上样于质量分数7.5% SDS-PAGE凝胶泳道中,80~120 V电压下电泳1.5 h,恒定电流0.36 A冰浴1.5 h转印至PVDF膜;将PVDF膜置于含质量分数5%脱脂奶粉TBST中,室温下摇床上封闭1.5 h;TBST漂洗3次,每次15 min,根据蛋白分子大小裁膜,分别加入抗eNOS(1∶ 200)、p-eNOS(1∶ 200)、p-Akt(1∶ 1 000)、Akt(1∶ 1 000)、β-actin(1∶ 1 000)一抗,4 ℃孵育过夜;TBST洗膜3次后,加入HRP标记二抗(1∶ 5 000),室温下摇床上孵育1.5 h;TBST洗膜3次后,采用超敏型ECL发光法显影。采用ImageJ软件测定各条带灰度值,以β-actin为内参照,计算各蛋白相对表达水平。
1.3 统计学方法
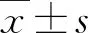
2 结果
2.1 各组大鼠造模后一般情况及不同时间点血糖浓度比较
5只模型大鼠血糖不达标被剔出实验,最终成功造模25只,糖尿病模型组及tBHQ干预组分别为15只和10只,饲养过程中未出现死亡大鼠。造模后72 h,糖尿病模型组大鼠已开始出现尿量增多、饮水增加的变化,随着病程延长,出现毛发粗糙、枯黄,精神萎靡,反应迟钝,多食、多尿、多饮症状明显;而tBHQ干预组大鼠多食、多尿、多饮症状较糖尿病模型组大鼠轻,精神状况较好,伴有一定程度的毛发粗糙和枯黄;正常对照组大鼠毛发顺滑、精神佳、反应灵敏。
造模后72 h、2周和4周各组间大鼠血糖浓度总体比较差异均有统计学意义(F=100.80,P<0.01;F=77.38,P<0.01;F=73.94,P<0.01)。糖尿病模型组造模后72 h、2周和4周大鼠血糖均较正常对照组及tBHQ干预组升高,差异均有统计学意义(均P<0.01)(表1)。
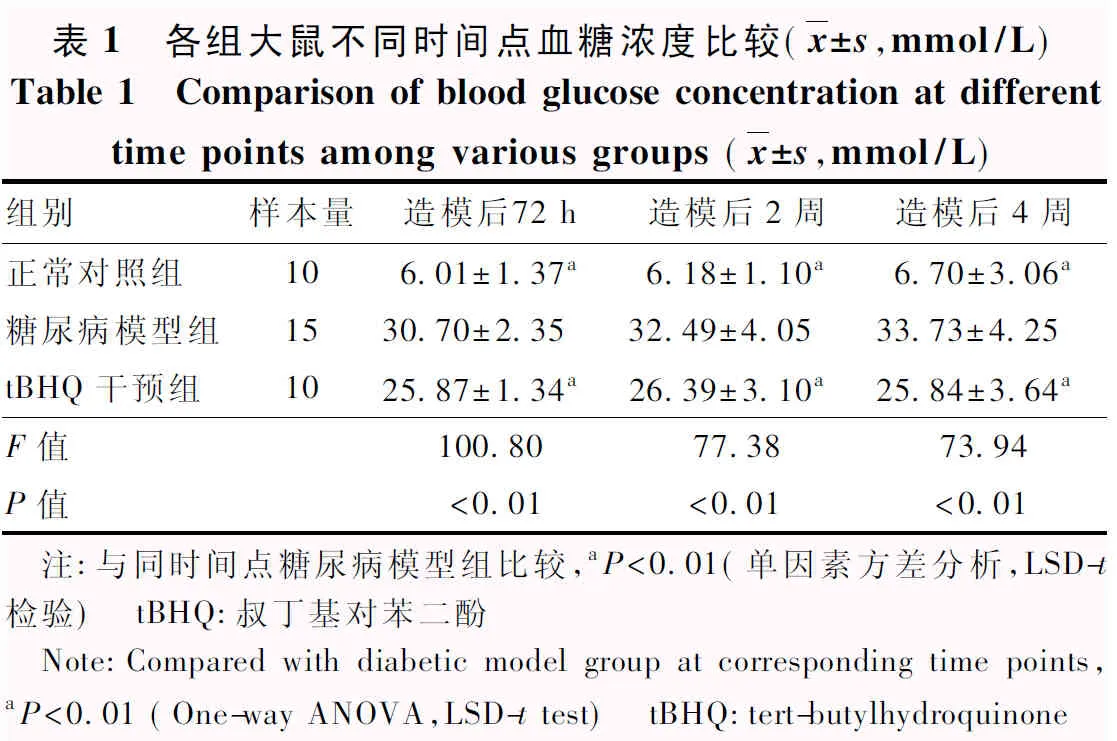
表1 各组大鼠不同时间点血糖浓度比较(x±s,mmol/L)Table 1 Comparison of blood glucose concentration at differenttime points among various groups (x±s,mmol/L)组别样本量造模后72 h造模后2周造模后4周正常对照组106.01±1.37a6.18±1.10 a6.70±3.06 a糖尿病模型组1530.70±2.3532.49±4.0533.73±4.25tBHQ干预组1025.87±1.34a26.39±3.10a25.84±3.64aF值100.8077.3873.94P值<0.01<0.01<0.01 注:与同时间点糖尿病模型组比较,aP<0.01(单因素方差分析,LSD-t检验) tBHQ:叔丁基对苯二酚 Note:Compared with diabetic model group at corresponding time points,aP<0.01 (One-way ANOVA,LSD-t test) tBHQ:tert-butyl-hydroquinone
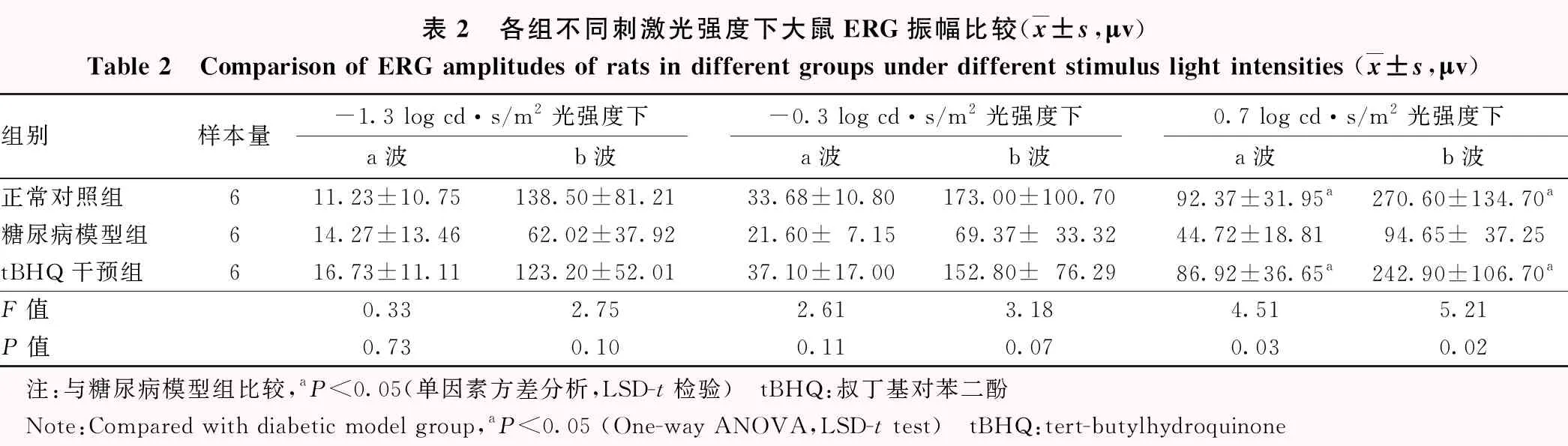
表2 各组不同刺激光强度下大鼠ERG振幅比较(x±s,μv)Table 2 Comparison of ERG amplitudes of rats in different groups under different stimulus light intensities (x±s,μv)组别样本量-1.3 log cd·s/m2光强度下-0.3 log cd·s/m2光强度下0.7 log cd·s/m2光强度下a波b波a波b波a波b波正常对照组611.23±10.75138.50±81.2133.68±10.80173.00±100.7092.37±31.95a270.60±134.70a糖尿病模型组614.27±13.4662.02±37.9221.60±7.1569.37±33.3244.72±18.8194.65±37.25tBHQ干预组616.73±11.11123.20±52.0137.10±17.00152.80±76.2986.92±36.65a242.90±106.70aF值0.332.752.613.184.515.21P值0.730.100.110.070.030.02 注:与糖尿病模型组比较,aP<0.05(单因素方差分析,LSD-t检验) tBHQ:叔丁基对苯二酚 Note:Compared with diabetic model group,aP<0.05 (One-way ANOVA,LSD-t test) tBHQ:tert-butylhydroquinone
2.2 各组大鼠ERG振幅比较
当光强度为-1.3 log cd·s/m2和-0.3 log cd·s/m2时,各组大鼠暗适应ERG的a波和b波振幅总体比较差异均无统计学意义(均P>0.05);当刺激光强度为0.7 log cd·s/m2时,各组大鼠暗适应ERG的a波和b波振幅总体比较差异均有统计学意义(F=4.51,P=0.03;F=5.21,P=0.02),糖尿病模型组暗适应ERG的a波和b波振幅明显低于正常对照组和tBHQ干预组,差异均有统计学意义(均P<0.05)(表2)。
2.3 各组大鼠视网膜结构比较
光学显微镜下可见正常对照组大鼠视网膜各层结构清晰完整,内界膜表面光滑,细胞排列整齐。糖尿病模型组大鼠视网膜组织层次结构尚完整但欠清晰,细胞间质水肿,内界膜表面粗糙,神经节细胞层细胞数量明显减少,内核层及外核层变薄、结构疏松且细胞排列紊乱。tBHQ干预组大鼠视网膜组织结构较完整,内界膜轻度增厚,神经节细胞层细胞轻度减少,内核层及外核层结构较疏松、细胞排列轻度紊乱(图1)。

图1 各组大鼠视网膜组织病理染色变化(HE ×400,标尺=50 μm) A:正常对照组大鼠视网膜各层结构清晰完整 B:糖尿病模型组大鼠视网膜层次欠清晰,各层细胞数量明显减少,层间结构疏松紊乱 C:tBHQ干预组大鼠视网膜结构基本完整,各层细胞数量轻度减少,层间结构较疏松 (箭头示内界膜) GCL:神经节细胞层;INL:内核层;ONL:外核层Figure 1 Histopathology of rat retinal tissues (HE ×400,bar=50 μm) A:The retinal layer structures of normal control group were clear and complete B:The retinal layers were not clear with the number of cells in each layer significantly reduced,and the interlayer structure was loose and disordered in diabetic model group C:The retinal structure was basically complete,and the number of cells in each layer was slightly reduced,and the interlayer structure was slightly loose in tBHQ intervention group (Arrows indicated the inner limiting membrane)GCL:ganglion cell layer;INL:inner nuclear layer;ONL:outer nuclear layer
2.4 各组大鼠视网膜组织中细胞凋亡情况比较
正常对照组大鼠视网膜各层可见少量TUNEL红色荧光染色的凋亡细胞;糖尿病模型组大鼠视网膜中红色荧光染色细胞较正常对照组增多,主要位于外核层;tBHQ干预组大鼠视网膜中红色荧光染色细胞较糖尿病模型组减少(图2)。正常对照组、糖尿病模型组和tBHQ干预组细胞凋亡指数分别为(7.63±6.49)%、(55.26±26.63)%、(8.59±7.98)%,总体比较差异有统计学意义(F=10.04,P=0.01),糖尿病模型组细胞凋亡指数较正常对照组和tBHQ干预组显著升高,差异均有统计学意义(P=0.02、P<0.01)(图3)。

图2 各组大鼠视网膜组织细胞TUNEL染色情况(×200,标尺=75 μm) A:正常对照组大鼠视网膜组织中可见少量红色荧光染色细胞 B:糖尿病模型组大鼠视网膜外核层可见大量红色荧光染色细胞 C:tBHQ干预组大鼠视网膜外核层可见少量红色荧光染色细胞 (箭头示红色与蓝色荧光重合部分) GCL:神经节细胞层;INL:内核层;ONL:外核层Figure 2 TUNEL staining of rat retinal tissues (×200,bar=75 μm) A:Red fluorescent cells were rarely observed in rat retinal tissue of normal control group B:A large number of red fluorescent cells were observed in the outer nuclear layer of retina in diabetic model group C:Few red fluorescent cells were observed in the outer nuclear layer of retina in tBHQ intervention group (Arrows indicated overlapped red and blue fluorescence) GCL:ganglion cell layer;INL:inner nuclear layer;ONL:outer nuclear layer
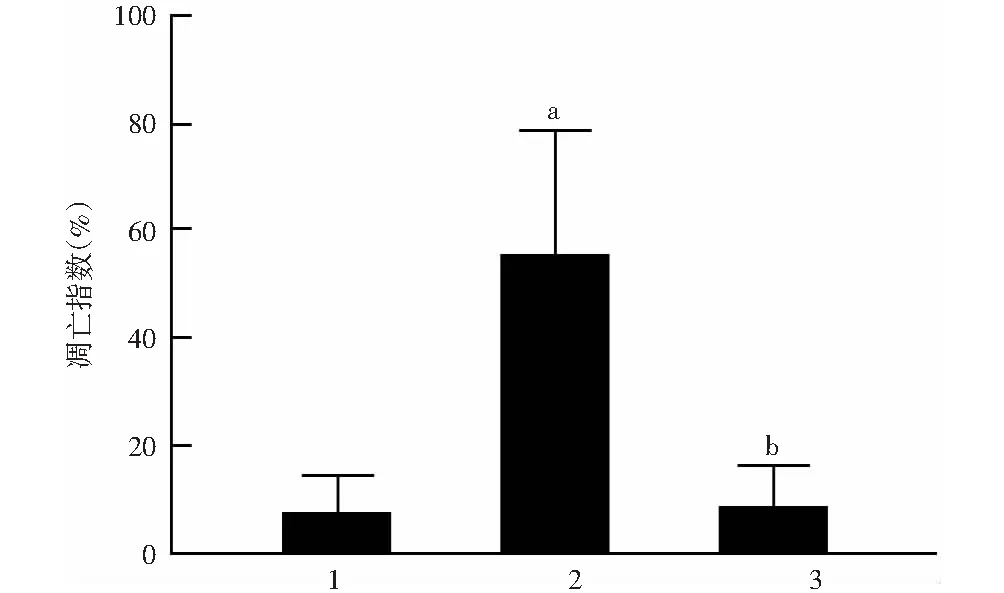
图3 各组大鼠视网膜组织细胞凋亡指数比较 F=10.04,P=0.01.与正常对照组比较,aP<0.05;与糖尿病模型组比较,bP<0.01(单因素方差分析,LSD-t检验,n=5) 1:正常对照组;2:糖尿病模型组;3:tBHQ干预组Figure 3 Comparison of apoptosis index of rat retinal tissue among different groups F=10.04,P=0.01.Compared with normal control group,aP<0.05;compared with diabetic model group,bP<0.01 (One-way ANOVA,LSD-t test,n=5) 1:normal control group;2:diabetic model group;3:tBHQ intervention group
2.5 各组大鼠视网膜组织及细胞中蛋白表达情况比较
正常对照组、糖尿病模型组、tBHQ干预组大鼠视网膜组织p-Akt/Akt的相对表达量分别为0.76±0.11、0.52±0.10和1.14±0.31,p-eNOS/eNOS的相对表达量分别为0.83±0.06、0.52±0.08和1.03±0.13,总体比较差异均有统计学意义(F=12.77,P<0.01;F=36.88,P<0.01);与正常对照组大鼠比较,糖尿病模型组大鼠视网膜组织中p-Akt/Akt和p-eNOS/eNOS相对表达量降低,差异均有统计学意义(均P<0.01);与糖尿病模型组比较,tBHQ干预组大鼠视网膜组织中p-Akt/Akt和p-eNOS/eNOS相对表达量显著升高,差异均有统计学意义(均P<0.01)(图4)。

图4 各组大鼠视网膜组织中p-Akt、Akt、p-eNOS和eNOS蛋白表达比较 A:各组p-Akt、Akt蛋白电泳条带 B:各组p-Akt/Akt蛋白相对表达量比较 F=12.77,P<0.01.与正常对照组比较,aP<0.01;与糖尿病模型组比较,bP<0.01(单因素方差分析,LSD-t检验,n=9) C:各组p-eNOS、eNOS蛋白电泳条带 D:各组p-eNOS/eNOS蛋白相对表达量比较 F=36.88,P<0.01.与正常对照组比较,aP<0.01;与糖尿病模型组比较,bP<0.01(单因素方差分析,LSD-t检验,n=9) 1:正常对照组;2:糖尿病模型组;3:tBHQ干预组 Akt:蛋白激酶B;eNOS:内皮型一氧化氮合酶Figure 4 Comparison of p-Akt,Akt,p-eNOS and eNOS protein expression in rat retinal tissues among different groups A:Protein bands of p-Akt and Akt B:Comparison of the relative expression of p-Akt/Akt protein F=12.77,P<0.01.Compared with normal control group,aP<0.01;compared with diabetic model group,bP<0.01 (One-way ANOVA,LSD-t test,n=9) C:Protein bands of p-eNOS and eNOS D:Comparison of the relative expression of p-eNOS/eNOS protein F=36.88,P<0.01.Compared with normal control group,aP<0.01;compared with diabetic model group,bP<0.01 (One-way ANOVA,LSD-t test,n=9) 1:normal control group;2:diabetic model group;3:tBHQ intervention group Akt:protein kinase B;eNOS:endothelial nitric oxide synthase
正常对照组、甘露醇对照组、高糖组、tBHQ干预组和PI3K抑制剂组细胞p-Akt/Akt的相对表达量分别为0.95±0.38、0.94±0.27、0.33±0.25、1.32±0.37和0.24±0.09,p-eNOS/eNOS的相对表达量分别为0.86±0.11、0.74±0.29、0.45±0.29、1.28±0.22和0.73±0.29,总体比较差异均有统计学意义(F=12.36,P<0.01;F=7.35,P<0.01);与正常对照组比较,高糖组细胞中p-Akt/Akt和p-eNOS/eNOS蛋白相对表达量明显降低,差异均有统计学意义(均P=0.02);与高糖组比较,tBHQ干预组细胞中p-Akt/Akt和p-eNOS/eNOS相对表达量明显升高,差异均有统计学意义(均P<0.01);与tBHQ干预组比较,PI3K抑制剂组中p-Akt/Akt和p-eNOS/eNOS相对表达量明显降低,差异均有统计学意义(均P<0.01)(图5)。

图5 各组细胞中p-Akt、Akt、p-eNOS、eNOS蛋白表达比较 A:各组p-Akt、Akt蛋白电泳条带 B:各组p-Akt/Akt蛋白相对表达量比较 F=12.36,P<0.01.与正常对照组比较,aP<0.05;与高糖组比较,bP<0.01;与tBHQ干预组比较,cP<0.01(单因素方差分析,LSD-t检验,n=9) C:各组p-eNOS、eNOS蛋白电泳条带 D:各组p-eNOS/eNOS蛋白相对表达量比较 F=7.35,P<0.01.与正常对照组比较,aP<0.05;与高糖组比较,bP<0.01;与tBHQ干预组比较,cP<0.01(单因素方差分析,LSD-t检验,n=9) 1:正常对照组;2:甘露醇对照组;3:高糖组;4:tBHQ干预组;5:PI3K抑制剂组 Akt:蛋白激酶B;eNOS:内皮型一氧化氮合酶Figure 5 Comparison of p-Akt,Akt,p-eNOS and eNOS protein expression in rMC-1 cell among different groups A:Protein bands of p-Akt and Akt B:Comparison of the relative expression of p-Akt/Akt protein F=12.36,P<0.01.Compared with normal control group,aP<0.05;compared with high glucose group,bP<0.01;compared with tBHQ intervention group,cP<0.01 (One-way ANOVA,LSD-t test,n=9) C:Protein bands of p-eNOS and eNOS D:Comparison of the relative expression of p-eNOS/eNOS protein F=7.35,P<0.01.Compared with normal control group,aP<0.05;compared with high glucose group,bP<0.01;compared with tBHQ intervention group,cP<0.01 (One-way ANOVA,LSD-t test,n=9) 1:normal control group;2:mannitol control group;3:high glucose group;4:tBHQ intervention group;5:PI3K inhibitor group Akt:protein kinase B;eNOS:endothelial nitric oxide synthase
3 讨论
既往研究表明,tBHQ具有保护STZ诱导的糖尿病模型大鼠视网膜的作用,并在一定程度上降低模型大鼠的血糖,增加视网膜组织中HO-1、Bcl-2等抗凋亡及抗氧化应激因子的表达[9-10]。本研究结果也显示,tBHQ干预的糖尿病大鼠血糖较糖尿病模型组显著降低,证实了tBHQ具有对抗高血糖的作用,其机制可能是tBHQ发挥了对糖尿病模型大鼠胰岛β细胞的保护作用,tBHQ干预的糖尿病模型大鼠可分泌更多的胰岛素,从而达到抗高血糖的作用[11]。
临床研究表明,DR患者出现眼底微血管病变之前视网膜功能已出现异常[12],主要表现为色觉、对比敏感度以及暗适应能力的降低,ERG表现为振幅下降及潜伏期延长[13]。动物实验也显示糖尿病大鼠发生视网膜功能损害早于血管病变[14]。ERG a波和b波主要来源于视网膜光感受器细胞(视锥细胞、视杆细胞)及双极细胞,反映细胞受到刺激时的电位变化。本研究结果显示,造模后4周糖尿病模型组大鼠ERG出现异常,与正常对照组相比主要表现为a波和b波振幅下降,说明STZ诱导的糖尿病大鼠早期即出现ERG振幅下降,与Chesler等[15]研究发现的糖尿病模型大鼠早期出现视功能障碍的结果一致。有证据表明,tBHQ在神经退行性病变中具有潜在的皮质神经元及运动神经元的保护作用[16]。本研究结果显示,tBHQ干预组大鼠ERG的a波和b波振幅较糖尿病模型组有所升高,推测tBHQ可在一定程度上改善糖尿病模型大鼠的视网膜功能,这种作用可能是tBHQ通过保护糖尿病模型大鼠的视网膜神经细胞产生的。
本研究中组织病理染色结果显示糖尿病模型组大鼠视网膜外核层及内核层变薄、细胞排列紊乱;TUNEL染色结果显示糖尿病模型组大鼠视网膜中凋亡细胞主要位于外核层,推测已有光感受器细胞和双极细胞受到损害,并且细胞数量减少。而tBHQ干预组大鼠视网膜上述情况则有所改善,说明tBHQ对糖尿病大鼠视网膜结构及神经细胞损害均有保护作用。各组大鼠视网膜组织形态结构特征及细胞凋亡情况也与ERG a波和b波振幅改变一致。
高糖诱导的氧化应激被认为是导致DR发生和发展的关键机制之一[17]。因此各种抑制氧化应激反应的蛋白或细胞因子对防治DR进展具有积极作用。eNOS作为机体内催化NO合成的酶之一,其在DR发病机制中有重要作用。病理情况下,eNOS来源的NO与超氧阴离子(O2-)反应,形成过氧亚硝酸盐(ONOO-),诱导细胞发生氧化应激反应,进而发生细胞凋亡,同时增强血管通透性[18];大量的氧化产物可导致eNOS解偶联及失活,导致氧化应激的恶性循环[19]。本研究发现,糖尿病模型大鼠视网膜组织中及高糖培养的细胞中磷酸化eNOS表达下降,提示高糖状态下eNOS存在失活,可能介导DR过程中高糖导致的视网膜结构和功能损害;在使用tBHQ干预的糖尿病大鼠视网膜或高糖细胞中发现,磷酸化eNOS表达升高。Chen等[20]研究表明,eNOS ser1179位点磷酸化能提高eNOS对辅因子(L-精氨酸、BH4)的亲和力,改善病理情况下的eNOS解偶联,减少eNOS失活,提示tBHQ具有促进或恢复eNOS活性的作用,并可能由此发挥抗视网膜神经元凋亡及抗氧化应激的作用。
eNOS的上游信号通路PI3K/Akt可介导人类多种疾病,包括癌症、糖尿病、心血管疾病和神经疾病[21-22]。既往研究表明,Akt ser473位点被磷酸化后,可激发Akt的完全酶活性,通过抑制促凋亡蛋白(如Bad)或抑制促凋亡信号(Foxo)实现调节作用[23]。在DR的发病过程中,PI3K/Akt通路被抑制,Akt磷酸化表达水平显著降低,下游凋亡级联反应被激活,引起视网膜神经元的氧化应激损伤和凋亡[24]。本研究中结果显示,糖尿病大鼠视网膜组织和高糖培养的细胞中磷酸化Akt表达降低,而tBHQ干预的糖尿病大鼠视网膜或高糖细胞中磷酸化Akt表达升高,表明tBHQ可能通过促进Akt蛋白ser473位点的磷酸化,使Akt活化,发挥对DR中视网膜的保护作用。进一步探讨tBHQ的保护作用机制发现,PI3K抑制剂组细胞中磷酸化Akt和磷酸化eNOS表达水平较tBHQ干预组下降,表明tBHQ的促进Akt和eNOS磷酸化作用可被PI3K抑制剂阻断,但其确切的机制有待进一步研究。
综上所述,本研究结果表明tBHQ可能通过PI3K活化Akt/eNOS途径对早期糖尿病模型大鼠视网膜结构和功能发挥保护作用,为tBHQ及其衍生制品防治DR视网膜损伤提供了理论依据,对早期DR的防治具有重要意义。但因为视网膜结构的复杂性及细胞的多样性,本研究中未对视网膜细胞间相互作用进行实验,并且尚缺乏tBHQ通过Akt介导的eNOS ser1179磷酸化的直接证据,有待后续的研究进一步探讨。
利益冲突所有作者均声明不存在利益冲突
作者贡献声明王娟:参与选题与实验设计、实验操作、数据整理、统计分析、论文撰写及修改;韦芳:参与实验设计、实验指导与实验操作;曹阳、田敏:参与实验选题、实验设计、实验指导;吕红彬:参与实验选题、实验指导、论文修改及定稿
猜你喜欢
中风与神经疾病杂志(2022年9期)2022-10-19
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
家禽科学(2021年10期)2021-11-22
现代临床医学(2021年5期)2021-11-02
天津医科大学学报(2021年4期)2021-08-21
安徽医药(2021年4期)2021-04-09
世界中医药(2019年11期)2019-09-10
中国现代医生(2019年4期)2019-04-10
湖南中医药大学学报(2018年9期)2018-09-10
